Review Article - Imaging in Medicine (2012) Volume 4, Issue 4
Cord blood endothelial progenitor cells as therapeutic and imaging probes
Branislava Janic1* and Ali S Arbab1
1Cellular & Molecular Imaging Laboratory, Department of Radiology, Henry Ford Hospital, 1 Ford Place, 2F, Box 82, Detroit, MI 48202, USA
- *Corresponding Author:
- Branislava Janic
Cellular & Molecular Imaging Laboratory
Department of Radiology, Henry Ford Hospital
1 Ford Place, 2F, Box 82, Detroit, MI 48202, USA
Tel: +1 313 874 1681
Fax:+1 313 874 4494
E-mail: bjanic@rad.hfh.edu
Abstract
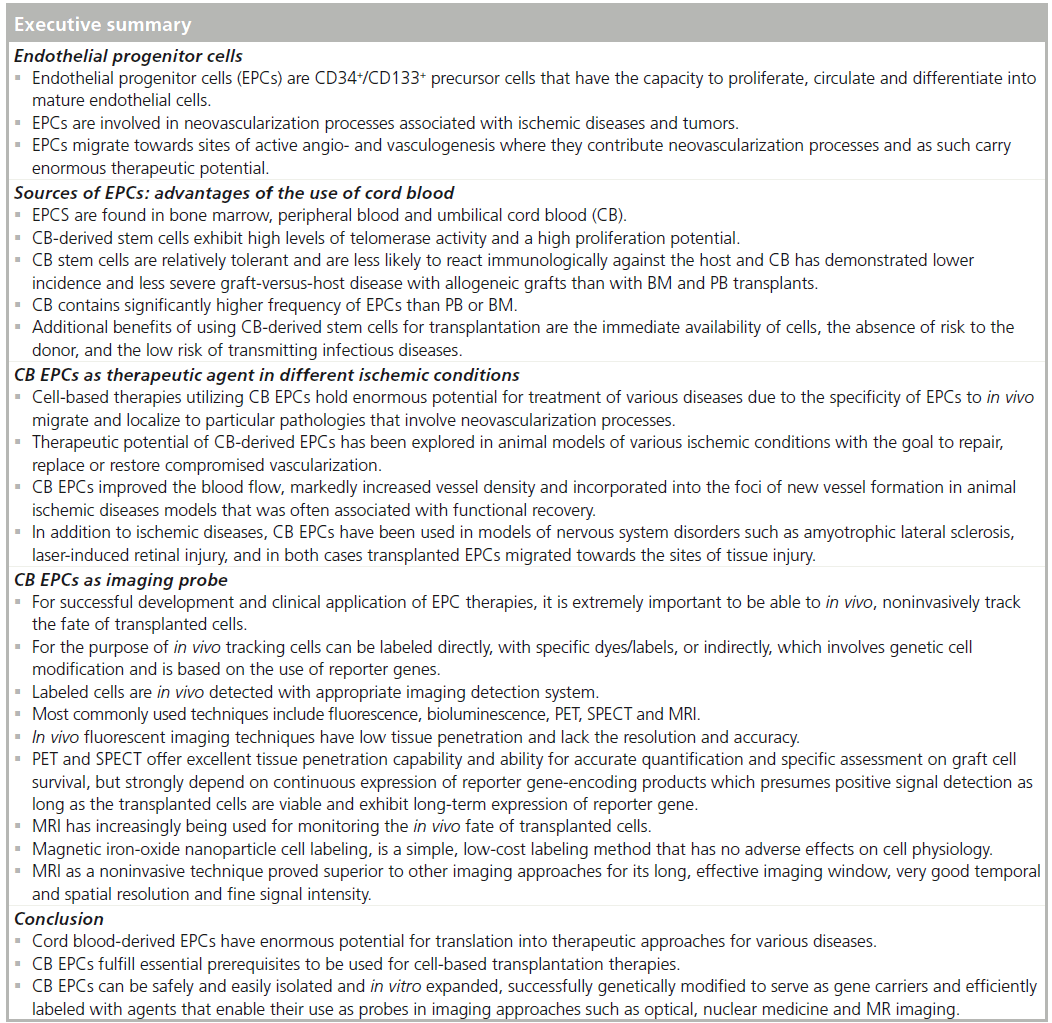
Numerous studies demonstrated that neovascularization processes associated with severe tissue ischemia commonly found in conditions such as cardiovascular disorders and tumor growth occur via angiogenic and vasculogenic mechanisms. Over the past decade, it has been demonstrated that endothelial progenitor cells (EPCs) play a significant role in neo-angiogenic and neovasculogenic processes. Due to their ability to self-renew, circulate, home to the ischemic sites and differentiate into mature endothelial cells, EPCs derived from various sources hold enormous potential to be used as therapeutic agents in pro- or anti-angiogenic strategies for the treatment of ischemic and tumor conditions, respectively. However, the development of EPC-based therapies requires accompanying, noninvasive imaging protocol for in vivo tracking of transplanted cells. Hence, this review focuses on cord blood-derived EPCs and their role in neovascularization with emphasis on the potential use of EPCs as a therapeutic and imaging probe.
Keywords
angiogenesis; cord blood; endothelial progenitor cells; imaging probe; vasculogenesis
The formation of blood vessels occurs by two mechanisms: vasculogenesis and angiogenesis. Vasculogenesis is the process where blood vessels are formed de novo by in situ differentiation of the primitive progenitors, such as angioblasts, into mature endothelial cells (ECs), which was thought to only take place during embryonic development [1]. By contrast, angiogenesis occurs both during the embryonic development and the postnatal life, and is defined as a process that gives rise to new blood vessels by proliferation and migration of pre-existing, differentiated ECs [2]. These ECs contribute to neovasculatures by sprouting and co-option of neighboring pre-existing vessels [3]. However, there is emerging evidence indicating that bone marrow-derived endothelial progenitor cells (EPCs) also contribute to the vasculogenesis and growth of certain tumors [4]. Early studies showed that EPCs can originate from a subpopulation of CD34+ human hematopoietic stem cells (HSCs) identified by the cell-surface molecule CD133, and these progenitors have been shown to be more specific for endothelial differentiation and angiogenesis [5,6]. In addition, other genes associated with angiogenesis regulation were observed to be highly expressed in CD133+ cells. EPCs collected from bone marrow (BM), peripheral blood (PB) or cord blood (CB) have been used in different animal tumor or ischemia models to determine whether these cells have the capacity to become part of the neovasculatures in tissues. When infused into immune-compromised mice, EPCs were incorporated into the vasculature of xenotransplanted tumors and were correlated to tumor volume and production of VEGF [7]. Additionally, our published results also showed migration and incorporation of intravenously administered CD34+/CD133+ cells in the xenoplanted tumors’ neovasculatures [8]. As reported by our group and other investigators, hypoxiainduced SDF-1a has been detected as one of the chemo-attractants that is crucial for migration and vascular incorporation of BM-derived EPCs owing to the presence of CXCR4 receptors in these cells [9,10]. Moreover, we have also reported the role of inflammatory cytokine RANTES, which also act as a chemo-attractant for these EPCs [6].
The capacity of EPCs or CD34+/CD133+ stem cells to migrate and incorporate into neovasculatures is usually demonstrated by histopathology or immunohistochemistry. However, there is a need to develop techniques that would enable in vivo, noninvasive imaging and tracking of transplanted cells. Recently it was demonstrated that MRI can monitor the migration of systemically administered magnetically (iron labeled) labeled mammalian cells to different organs, tumors and other sites of active angiogenesis [11]. Nuclear medicine and optical imaging techniques are also being used to determine the migration and accumulation of administered cells to the sites of lesion or sites of interest.
The potential use of stem cell-based therapies for the repair and regeneration of various tissues and organs offers alternative therapeutic solutions for a number of diseases. Reports by US Department of Health and Human Services stated that almost one in three individuals in the USA could potentially benefit, over their lifetime, from regenerative medicine. Replacing or regenerating affected tissues with the use of stem/progenitor cells holds the promise to greatly affect the outcome in many conditions that were, in most cases, treated with palliative methods. However, choosing the correct source and type of stem/progenitor cell and generating a sufficient number of cells, as well as in vivo monitoring of biodistribution of transplanted cells are critical factors in determining the success of stem/progenitor cell-based therapies. EPCs represent a minor population of precursor cells that can be found within mononuclear cells (MNCs) in BM, PB and CB. Since the discovery that EPCs may play an important role in vasculogenesis and angiogenesis, these cells have become the focus of developing cell therapy approaches for various conditions, including vascular ischemic diseases and tumors. Under ischemic conditions, EPCs migrate and accumulate to the sites of hypoxia where they integrate and contribute to the ongoing neovascular processes. EPCs can be used either to enhance the repair of diseased, ischemic tissues by enhancing and/or participating in neovascularization or as a gene carrier with the purpose of inhibiting the growth of a particular tumor. In this review, we will discuss EPCs, their sources, characteristics and potential use as imaging and therapeutic probes.
Endothelial progenitor cells
EPCs are precursor cells that have the capacity to proliferate, circulate and differentiate into mature ECs. EPCs were implicated to play a critical role in adult, postnatal endothelial repair and vasculogenesis in conditions such as myocardial ischemia and infarction, unstable angina, stroke, limb ischemia, wound healing, atherosclerosis, diabetic microvasculopathies, ischemic retinopathies, pulmonary arterial hypertension and tumor vascularization [12–17]. Our group has performed extensive in vitro and in vivo studies that demonstrated the endothelial potential of CD34+/CD133+ cells. Our recent publication showed the potential of long- and short-term cultured CB-derived CD133+ EPCs to make tube-like structures both in vitro and in vivo matrigel angiogenesis studies [6]. In addition, some of our in vivo animals studies showed the incorporation of administered CD133+ EPCs in the tumor neovasculatures [8,10]. Altogether, these findings granted significant therapeutic potential and EPCs became one of the main focuses of regenerative medicine. Pioneering studies on EPCs demonstrated that CB, BM and PB can serve as the enriched source of CD34+/CD133+ EPCs [18,19]. However, these cells were initially characterized as hematopoietic stem/progenitor cells and since then, EPCs have been a controversial topic and focus of continuous research effort to unambiguously delineate the source, phenotype and differentiation hierarchy of the putative EPCs. The first studies defined EPCs as the cells co-expressing hematopoietic stem cell (HSC) marker CD34, and EC marker VEGFR2 [19]. However, it has been demonstrated that some mature ECs also co-express CD34 and VEGFR2. In addition, CD34 was also shown to be expressed on various nonhematopoietic populations. Therefore, there was a need for characterizing a more specific EPC marker. Subsequent studies focused on a novel, highly conserved CD133 glycoprotein, with unknown biological activity that was expressed on HSCs but not on mature ECs, and therefore was acknowledged as a more suitable marker for immature progenitor cells [5,20–22]. Furthermore, it was suggested that a subset of circulating CD34+ cells that express both VEGFR-2 and CD133 represents a functional EPC population that plays a role in postnatal angiogenesis or vasculogenesis [20]. Some investigators pointed out that EPCs should be defined as CD45 negative and should express CD31, VEGFR2, VE-cadherin and vWF [23,24] It is generally accepted that CD133+/CD34+/VEGFR2+ cells represent an immature, highly proliferative EPC population that gives rise to more mature CD133-/CD34+/ VEGFR2+ cells that are limited in their proliferative capacity [12], and this proliferative aspect is extremely important for developing future therapies. Currently, EPCs are defined as cells that are positive for CD133, CD34 and VEGFR2 markers. In addition, these cells also express some of the endothelial-specific antigens such as platelet EC adhesion molecule 1 (PECAM-1 or CD31), E-selectin and VE-cadherin, as well as the chemokine receptor CXCR-4 (CD184), and have the ability to migrate in response to the CXCR-4 ligand, SDF-1. During differentiation into mature ECs, expression of CD34 and CD133 is downregulated [22,25] and continuous culturing of EPCs increases the expression of mature EC markers [26]. On the other hand, some reports demonstrated that CD45 positive cells exhibited angiogenic potential as well as all the hallmarks of typical EPCs [27,28]. Despite the significant effort to characterize EPCs’ phenotypical and functional, properties overlap among EPCs, HSCs and ECs still exists and remains the source of controversy. It is still not clear whether EPCs’ therapeutic potential is contingent on differentiation/maturation status. Our work is focused on EPCs that we define as subpopulation of CD34+ cells that are CD133 positive (CD34+/CD133+), and therefore these cells are the focus of this review.
Although initial clinical trials in patients with ischemic limb or heart disease indicated the feasibility of safe and effective use of EPCs transplants [29–35], the successful translation of EPC therapy is still limited by low quantities of EPCs that can be generated from patients. Moreover, it has been shown that in vivo numbers and percentage of functional EPCs decreases with aging [36] and in conditions such as cardiovascular diseases [37,38]. Therefore, to further enhance therapeutic efficacy and avoid poor therapeutic responses due to EPCs’ impairment, it is important to develop strategies to expand, preserve and have readily available ex vivo a sufficient number of functional EPCs. Several different methodological approaches for ex vivo EPC enhancement and amplification have been reported. However, different culture conditions, such as suspension versus adherent culture with various cytokine combinations, generated EPCs exhibiting significant differences with regard to phenotypes, function and, very often, proliferative potential as well [5,23]. Initially, the majority of groups employed methods that were based on enriching for attached endothelial lineage cells by culturing PB-, BM- or CB-derived mononuclear cells in the presence of VEGFs [23,39] and these methods generated cells that exhibited vasculogenic properties and functional recovery in in vivo animal models [40–42]. However, the in vitro expansion of attached EPCs phenotype was associated with increase in senescence and inferior proliferation capacity [43]. Therefore, several culture systems have been developed employing a suspension type of in vitro expansion that generated functional EPCs [40,42–45]. Our group has recently reported a culture system that utilized 30‑day ex vivo expansion of CB-derived CD133+ EPCs in serum-free media conditioned with stem cell factor (SCF), FLT3 and thrombopoietin (TPO) [6]. Cells cultured under these conditions achieved 148-fold increases in cell numbers. When induced to differentiate, they expressed VE-Cadherin, VEGFR2, vonWillebrand factor, CD31, CD54, CD184, CD62E, CD29, CD150, CD195 and CD105, and were positive for acetylated low-density lipoprotein uptake. The cells also stimulated endothelial tube formation in co-culture with mature ECs and exhibited a migratory response to SDF-1a and RANTES. In addition, when we used these in vitro expanded CB CD133+ EPCs in tumor animal models, either locally [10] or systemically [8] administered, they migrated and incorporated into the tumor vasculatures. Altogether, our reports indicated that serum-free, suspension culture conditions facilitated ex vivo expansion of EPCs that preserved angiogenic properties under in vitro and in vivo conditions. Recently, similar results using parallel culture conditions for CB-derived CD34+ cells were reported by Ahrens et al. [44]. They also showed that under differentiation conditions these cells significantly upregulated genes associated with cardioprotective and pro-angiogenic properties.
Sources of EPCs: advantages of the use of CB
The three most used sources of EPCs are BM, mobilized PB and umbilical CB. BM-derived CD133+ cells improved tissue regeneration when autologously transplanted to myocardial infracted patients [46,47], while CB cells were used in a clinical trial of a variety of malignant and nonmalignant hematological disorders [48]. Since its initial clinical use in 1989, CB has been considered as an interchangeable alternative to BM and mobilized PB and as just another source of stem/progenitor cells. However, new evidence accumulated over the past decade demonstrates that umbilical CB provides distinct advantages over other EPC sources and has the potential to be therapeutically applied across a wide range of pathological conditions. Therefore, as a legitimate resource of stem cells, CB became an attractive choice for tissue engineering and regenerative medicine.
In terms of ontogeny, CB-derived stem cells are at the intermediate point between embryonic and adult life [49]. CB stem cells also exhibit longer telomeres associated with high levels of telomerase activity and a high proliferation potential [50–52]. It appears that CB stem cells are relatively tolerant and are less likely to react immunologically against the host [53,54]. Hematological transplantation using CB has demonstrated lower incidence and less severe graft-versus-host disease with allogeneic grafts than with BM and PB transplants, despite the HLA disparity [48,55,56], hence providing a significantly higher chance of finding a donor for populations who are under-represented in donor registries. Therefore, CB-derived cells may be very important as a transplant source for patients lacking appropriate adult donors. It has also been shown that CB contains a significantly higher frequency of EPCs than PB or BM [57]. Additional benefits of using CB-derived stem cells for transplantation are the immediate availability of cells, the absence of risk to the donor, and the low risk of transmitting infectious diseases (cytomegalovirus or Epstein–Barr virus) [54]. With the current global rate of approximately 100 million per year, CB remains the largest source of stem cells available and to date more than 450,000 unrelated units are banked worldwide and are available for potential clinical use [58]. In addition, the noninvasive nature of its collection and potential for easy and efficient characterization and banking grant CB-derived stem cells a unique therapeutic prospect [59].
CB EPCs as a therapeutic agent in different ischemic conditions
The therapeutic potential of CB-derived EPCs has been explored in numerous animal models. The majority of work with most promising results utilized these cells in various ischemic models with the goal to repair, replace or restore compromised vascularization in various tissues or organs. In animal models of hind limb ischemia, CB-derived EPCs were effective in the recovery of blood flow and neovascularization. Recent studies by O et al. demonstrated that when locally injected into the ischemic thigh muscle area, CB CD34+ EPCs improved the blood flow, markedly increased vessel density and incorporated into the foci of new vessel formation [45]. Cells used in these experiments were previously in vitro expanded in serum free suspension culture. Similar results were also reported from earlier studies using the same model, where CB EPCs were administered either locally [39] or systemically [60]; however, in both cases prior to injection the EPCs were ex vivo expanded as an attached cell population. To date there has been no report on whether angiogenic effect observed in animal models depends on the route of CB EPC administration. Considering the advantages demonstrated for CB over other sources of EPCs, promising results from clinical trials using peripheral blood CD34+ EPCs [29] strengthen the prospects of developing and clinically translating CB EPCs angiogenic therapies for critical limb ischemia.
Another ischemic condition that can significantly benefit from CB EPC-based therapy is myocardial infarction where physiological angiogenesis is unable to repair the injury. A significant body of work carried out on myocardial infarction animal models and clinical trials demonstrated the therapeutic potential of EPCs regardless of their source. However, the discovery that patients with cardiovascular disorders have decreased numbers of endogenous EPCs exhibiting greater in vitro senescence [36,37,61] underscores the importance of utilizing CB EPCs as an alternative nonautologous approach. Recent work using a rat model of myocardial infarction demonstrated that intracardiac implanted CB CD133+ EPCs significantly improved left ventricular ejection fraction as compared with the control group of rats [62]. However, the results were not further supported with analysis on the mechanisms underlying the observed functional improvement. On the other hand, in the similar rat model Schlechta et al. also used CB CD133+ EPCs but via systemic tail vein injection [40]. They observed an approximately 30% improvement 4 weeks after cell administration in left ventricular ejection fraction in treated animals, and this functional improvement was of higher magnitude than the reported improvement after local CB CD133+ EPC administration [62]. This difference in results reported by the two groups indicate that in myocardial infarction, systemic EPC administration may have the advantage over local intracardiac implantation, which in the clinical setting with already seriously compromised, infarcted patients, would be a less desirable route of administration. Nevertheless, following various reports on the effect of PB and BM EPCs on cardiac recovery in animal models of myocardial infarction or in clinical studies [30–33,63], both groups contributed in emphasizing the potential of CB EPCs in myocardial infarction functional recovery.
Cerebral ischemia is yet another ischemic condition where CB EPCs may have a great potential significance. Cerebrovascular diseases are the third leading cause of death in the USA, with approximately 700,000 people affected annually [64] and all the reported studies to date have indicated extreme potential/ role of stem cells for angiogenic and neurogenic recovery after cerebrovascular insults. Some of the first evidence came from the work by Chen et al., where they showed that in middle carotid artery occlusion (MCAO) rat stroke model injection of CB CD34+ cells reversed stroke-induced behavioral deficits [65]. However, later work in animal models mainly used either nonfractionated CB or mononuclear CB cells without further cell subtype enrichment. Nevertheless, accumulated evidence showed therapeutic benefit with regard to neuroprotective effect and functional, behavioral improvement [66–69]. One of the mechanisms behind the observed improvements included decrease in inflammation with reduction in tissue cytokine levels and number of infiltrating inflammatory cells, as well as a decrease in resident glial cell activation [67,68]. In addition, CB administration also reduced the ischemic volume [68,69], protected neuronal tissue from apoptosis and induced reorganization of nerve fibers [69]. It is likely that the combinations of effects observed are the result of the interactive play of different cell populations contained within the unfractionated, administered CB, and this multicellular type approach may be necessary for the reported effects to take place. Still, the latest studies have attempted to decipher the mechanisms when separate CB-cell populations were administered in animal stroke models. Nystedt et al. showed that human CB CD34+ cells slightly improved behavioral and sensorimotor functions in MCAO rats after systemic administration, but failed to provide neuroprotection and influence infarct volume [70]. A very recent study tried to shed light on the particular role of CB CD34+ in providing neuroprotection in a rodent MCAO model [71]. It has been shown that intravenous injection of CB CD34+ cells reduced neurofunctional deficit and diminished infarction volume. However, the magnitude of this effect was inferior compared with the effect of CB MNC fraction administered under the same conditions. The authors suggested that MNCs provided the most prominent neuroprotection with CD34+ cells playing a particular role in the MNCs protective action [71]. Further experimentation using animal stroke models are warranted to determine the effective cells within the heterogeneous CB population and their modes of action.
In addition to stroke, CB EPCs have been used in other models of nervous system disorders. These reports mainly demonstrated the ability of administered CB EPCs to migrate and reside at sites of injury. Willenbrock et al. injected magnetically labeled CB CD34+ cells intraspinally into a transgenic mouse model of amyotrophic lateral sclerosis [72]. CB EPCs were also used in laser-induced retinal injury in mouse and these experiments demonstrated the ability to in vivo monitor the fate of injected, fluorescently labeled cells. In addition, the administered cells incorporated into the neovasculature within the retinal tissue [73].
CB EPCs as an imaging probe
Cell-based therapies utilizing CB EPCs hold enormous potential for the treatment of various diseases owing to the specificity of EPCs to in vivo migrate and localize to particular pathologies that involve neovascularization processes. However, for the successful development and clinical application of such therapies, it is extremely important to be able to in vivo, noninvasively track the fate of transplanted cells. In addition, monitoring the delivery of the administered cells, their in vivo migration to the site of injury and subsequent in situ differentiation or proliferation would also enhance the understandings of the particular neovascularization mechanisms involved, and this can further help to enhance EPC therapy for the given disease. CB EPCs have been utilized in various medical imaging settings. Commonly, for the purpose of in vivo tracking these cells can be labeled directly, with specific dyes/labels, or indirectly, which involves genetic cell modification and is based on the use of reporter genes. In both cases, labeled cells are in vivo detected with an appropriate imaging detection system. The most commonly used techniques include fluorescence, bioluminescence, (PET), SPECT and MRI and we will briefly summarize the use of CB CD133+/CD34+ EPCs with these techniques.
CB EPCs in optical imaging
Fluorescence imaging techniques for labeling CB EPCs are largely used in cellular and developmental biology with the purpose of deciphering the mechanisms of EPC involvement in neovascularization. Most commonly, fluorescent cell labeling applications are based on the use of organic fluorophores such as rhodamine, fluorescein, alexa488 and other similar probes and are used for short-term in vivo tracking or for the purpose of histologically confirming tissue presence of labeled cells at the end point of the study [40]. Common problems associated with the use of these dyes in in vivo settings are photobleaching and quenching, and sensitivity to pH changes and chemical degradation. Nevertheless, several fluorescent agents, such as 1,1´-dilinoleyl-3,3,3´,3´-tetramethylindocarbocyanine perchlorate-labeled acetylated low-density lipoprotein (DiI-AcLDL), 5- (and-6-) -carboxyf luorescein diacetate succinimidyl ester (CFSE) and PKH26 have been frequently used in labeling stem/progenitor cells. Among the fluorochromes used for stem/progenitor cell labeling, CFSE stands out as the most suitable for long-term cell tracking and quantifying proliferation in vivo and in vitro [74–76]. DiI-AcLDL has been used to identify the EC population [77,78] and to assess ECs’ functional integrity [79,80], and is therefore extremely helpful in analyzing EPC differentiation towards EC lineage in the in vitro setting but may not be a good agent for the in vivo study. Recent work by Shi et al. utilized CFSE to in vivo track CB EPCs in mouse model of laser-induced retinal injury, and the fluorescent cells were identified around photocoagulated lesions 1 week after the transplantation of labeled EPCs. Furthermore, 4 weeks later neovascular incorporation of these cells was detected as fluorescent tube-like structures within the retinal vasculature [73].
One of the major alternative ways to ex vivo (i.e., in vitro) label cells is the use of reporter gene. One of those widely used in cell tracking, including EPCs, is green fluorescent protein (GFP) that has been shown to have better photostability and overall emission time than organic fluorophores [81,82]. Most often adenoand retro-viral vectors are used as a delivery mode for stably introducing GFP into the cells. However, vector-mediated gene transfer has proven difficult when transfecting stem cells, which was very often the reason why GFP+ EPCs isolated from transgenic GFP+ mouse were used as an alternative approach [83,84]. Our group has utilized the lentiviral reporter gene system to analyze the effectiveness of different promoters in driving the expression of GFP protein in CB CD133+ cells. We successfully achieved stable, long-term expression of the transduced GFP gene that was most robust in cells transfected with constructs containing cellular polypeptide chain EF1a and human cytomegalovirus (CMV) promoter [85]. A novel and interesting approach by Hayakawa et al. demonstrated the feasibility of long-standing, viral introduced GFP expression in CB CD34+ cells under in vivo condition [86]. Three months after injecting GFP transduced, human CB CD34+ cells into nonobese diabetic/severe combined immunodef icient NOD/SCID IL2Rγnull mouse, a significant (33%) percentage of peripheral blood cells were positive for the human CD45 marker, out of which 79.4% were GFP positive. In addition, cells harvested from the bone marrow of mice that received GFP+ CB CD34+ cells exhibited GFP expression in ex vivo culture for up to 18 days postplating [86]. Despite the efforts to optimize and improve techniques based on introducing and stably maintain GFP expression in transduced cells, drawbacks related to GFP detection and imaging still exist. For example, in in vivo models genetically encoded GFP exhibited spectral absorption and emission overlap with tissue autof luorescence with decrease in the contrast between the targets and the background. Moreover, the emission profile of GFP has limited tissue penetration capability making it less useful for in vivo imaging with regard to image resolution. However, developing dyes utilizing near infrared (NIR) range may circumvent problems encountered with GFP protein, since the absorbance spectra within NIR range has a minuscule overlap with absorbance properties of biomolecules within the tissue, as well as higher tissue penetration capability [87]. Recent findings from our group showed the ability of optical imaging (Kodak™ Multispectral system, Carestream Health Inc., NY, USA) to track the migration of GFP+ bone marrow cells to the sites of implanted tumors by in vivo imaging techniques [88]. We made a chimeric model by replacing the bone marrow of irradiated (6 Gy, whole body) host animals with GFP+ bone marrow from donor animals and allowing it to engraft for 4 weeks. After 4 weeks, more than 75% engraftment efficiency was achieved and the animal received either subcutaneous (breast cancer) or intracranial (glioma) implantation of tumors. In vivo imaging clearly showed the accumulation of GFP+ cells in the subcutaneously implanted breast cancer but in vivo detection of accumulation in the intracranial tumor was not conclusive. However, ex vivo optical imaging showed clear accumulation GFP+ cells in the brain tumors. This is our ongoing study to determine the percentage of BM EPCs accumulated in the tumors. Nevertheless, in vivo fluorescent imaging techniques still lack the resolution and accuracy necessary to accurately determine and quantify active cell migration and accumulation.
CB EPCs in nuclear medicine imaging
Nuclear medicine imaging techniques such as PET and SPECT offer many advantages over optical imaging such as excellent tissue penetration capability and ability for accurate quantification and specific assessment on graft cell survival. However, for in vivo cell tracking purposes these techniques strongly depend on continuous expression of reporter gene-encoding products, which presumes positive signal detection as long as the transplanted cells are viable and exhibit long-term expression of reporter gene. Long-term expression is usually achieved by viral expression systems, adenoand retro-virus vectors being the ones most commonly used. Adenovirus constructs usually deliver robust expression of reporter gene, but they may also exhibit expression leakiness, cause immunogenic reaction and not carry the expression to daughter cells [89,90]. Conversely, lentiviral vectors stably integrate into the host cell enabling long-term expression that carries into daughter cells and do not exhibit gene silencing and immunogenicity [91–93]. Regardless of the vector used, for the signal to be detected, a specific radioisotope or radiopharmaceutical probes need to be administered that will specifically interact with the reporter gene product. Gene reporter systems that are currently used in nuclear medicine imaging usually encode for a cell surface receptor that specifically binds the probe (dopamine D2 receptor), membraneassociated transporters that transport the probe across the cell membrane (sodium iodide symporter also known as NIS) or for enzymes that modify the probe (tk). In vivo CB EPC tracking by nuclear imaging has most effectively been achieved by using the hNIS reporter gene. hNIS is an intrinsic transmembrane glycoprotein that mediates transport of iodide into the thyroid follicular cells, and therefore is usually used in conjunction with radioactive iodine (such as I-123, I-124 or I-131) [94,95]. This active channel also transports technetium-99m (99mTc) pertechnetate (99mTc) that can be imaged by g camera [96,97]. Our group demonstrated the feasibility of transducing CB CD133+ EPCs with both, adeno- and retro-viral vectors. In a rat glioma model, we showed that intravenously administered CB CD133+ EPCs that were transduced with lentivirus carrying the hNIS gene, maintained the expression of this gene under in vivo conditions. This expression was detected by SPECT imaging as 99mTc activity and the data demonstrated that administered cells migrated to the tumor site and incorporated into the tumor neovasculature (Figure 1) [88]. Based on the percentage of accumulated In-111-labeled EPCs in the implanted glioma, we calculated that at least 1 × 105 NIS expressing EPCs were needed to be detected by 99mTc SPECT imaging [88]. Similar results were obtained from our earlier study on subcutaneous breast cancer model where we utilized CB CD133+ EPCs that were transfected with adenoviral vector carrying the hNIS gene [98]. In both studies we confirmed the results by using MRI to detect the same cells that were double labeled with iron oxides. A similar approach was reported by Higuchi et al. who used CB CD34+ EPCs that were retroviraly transduced with the NIS gene in rat model. These cells were locally implanted into the heart and 1–3 days later detected by 124I-PET imaging. However, when labeled with iron, the same cells were detectable by MRI up to 7 days after transplantation [99]. It is important to note that careful animal model selection is necessary to achieve the in vivo preservation of optimal numbers of viable, gene-expressing cells and to prevent the initiation of am immunogenic reaction and rejection of administered EPCs.
Nuclear medicine techniques were also utilized to track the IV administered radioisotope tagged CB EPCs in tumor neovascularization. Our group has reported the tracking of In-111- labeled CB EPCs in tumor neovascularization, which were also validated by MRI and histology (Figure 1) [100]. EPCs can easily be labeled with In-111-oxine by simple incubation for 25–30 min. Despite the efficacy of nuclear medicine in in vivo tracking, radioisotope-tagged cells, associated radiation injury and short halflife of available isotopes are the main drawbacks that needs to be overcome.
CB EPCs in MRI
Over the past decade MRI has increasingly being used for monitoring the in vivo fate of transplanted cells. The reasons are twofold: first, magnetic iron oxide nanoparticle cell labeling is a simple, low-cost labeling method that has no adverse effects on cell physiology [101], and therefore is slowly becoming a routine practice in cellular MRI. Second, MRI as a noninvasive technique proved superior to other imaging approaches for its long effective imaging window, very good temporal and spatial resolution and fine signal intensity [102]. To date, different studies have shown that various types of stem cells, when magnetically labeled with iron oxide nanoparticles, are detectable by MRI [103,104]. These nanoparticles have a high relaxation rate and they can shorten T2 relaxation time, which creates low signal intensity and therefore enables the area of interest in the given tissue to be visualized [105]. Our group has extensively used cellular MRI for in vivo tracking of CB CD133+ EPCs. We have developed and optimized labeling protocol that utilizes a superparamagnetic iron oxide nanoparticle ferumoxide, which we extensively used to label and track CB CD133+ EPCs in various animal models [106]. Our recent work using an angiogenesis model in nude mice demonstrated the presence of intravenously administered ferumoxide-labeled CB CD133+ EPCs within the newly grown vasculature within the matrigel implant. These cells were detected 7 days postinjection by ex vivo MRI conducted postmortem on collected tissues and tissue sections staining with Prussian Blue and they colocalized with newly formed vessel like structures [6]. To explore and decipher neoangiogenic processes and related molecular mechanisms, we have also utilized cellular MRI in tumor models. Cellular MRI of subcutaneous implants of ferumoxidelabeled CB CD133+ EPCs and melanoma or glioma cells in nude mice demonstrated hypointensities at tumor peripheries that colocalized with high expression of EC markers. These results demonstrated that migration of locally implanted, ferumoxide-labeled CB CD133+ EPCs to the sites of active angiogenesis at the tumor periphery can be monitored by cellular MRI [10]. We have also used cellular MRI as part of a multimodal approach in a glioma angiogenesis rat model, where we pointed out the difference in administered cell dose on detection threshold between SPECT and MRI. Our MRI results demonstrated that 7 days postintravenous injection, ferumoxide-labeled and hNIS-transduced CB CD133+ EPCs migrated to intracranial implanted glioma (Figure 2) . However, the SPECT analysis of the same did not detect sufficient amount of 99mTc when 5 million transgenic cells were administered but 99mTc SPECT clearly detected the migrated cells when the dose of transgenic cell numbers was increased to 10 million (Figure 2C–E) [88]. Wang et al. intravenously administered magnetically labeled CB EPCs in human glioma mouse model. Through cellular MRI they showed that administered cells home to the glioma site as early as 1 day after transplantation and these cells were mainly located at the periphery of the tumor where an abundant neovascular network was detected [107]. In addition to research on neoangiogenesis, CB EPCs in conjunction with cellular MRI were also used to study neurological disorders. Work by Willenbrock et al. successfully detected in vivo magnetically labeled intraspinally injected CB CD34+ EPCs in a transgenic model of amyotrophic lateral sclerosis. The persistent signal, with no reduction or dilution along the spinal cord, was detectable 4 days post-transplantation [72].

Figure 1. Migration and accumulation of administered In-111-oxine and SPIO-labeled endothelial progenitor cells in tumors. Five million In-111-labeled EPCs followed by 5 million magnetically labeled EPCs were administered in tumor-bearing rats. Both In-111- and SPIO-labeled EPCs were injected at the same time in the same animals. Following intravenous administration, SPECT images were obtained on days 0, 1 and 3. MRI images were obtained on day 7 to allow the decay of the radioactivity. SPECT images of the tumor obtained at 3 h (A), 24 h (B) and 72 h (C) showed increased activity at the site of tumor, indicating accumulation of In-111 labeled EPCs. Note a few of the iron-positive cells also make the lining of blood vessels (inset, black arrows). MRI was obtained by a clinical 3T system on day 7 following last SPECT. (D) T2-weighted image with an echo time of 35 ms, (E) T2-weighted image with an echo time of 20 ms and corresponding R2* map (F). Note the low signal intensity areas on T2-weighted image (E, black arrows) and corresponding R2* map (F, yellow arrows) indicating accumulation of iron-positive cells, which is proved by diaminobenzidine-enhanced Prussian blue staining (G). Inset showing the iron-positive cells lining the blood vessels. EPC: Endothelial progenitor cell. Reproduced with permission from [101].
Conclusion
CB-derived EPCs have enormous potential for translation into therapeutic approaches for various diseases. Over the last few decades, a significant body of work has generated evidence that CB EPCs fulfill essential prerequisites to be used for cell-based transplantation therapies. These cells can be safely and easily isolated and after in vitro expansion generate clinically relevant numbers. In addition, CB EPCs can be successfully genetically modified to serve as gene carriers; and lastly, they can be efficiently labeled with agents that enable their use as probes in imaging approaches such as optical imaging, nuclear medicine imaging and MRI.
Future perspective
Based on the availability of CB and placental tissues, CB will be one of the main sources to collect and bank pluripotent stem cells and different subpopulation of progenitor cells. With the established techniques of propagation, single subpopulation of progenitor cells then can be expanded and stored for future use. These stored cells then can be easily used in patients with various emergency conditions such as stroke, myocardial infarction and others. EPCs may be the main type of cells that can be used to salvage penumbra in stroke and ischemic myocardium, by generating new blood vessels. One advantage of CB-derived stem cells and EPCs would be to inject in a patient with a minor HLA mismatch (two HLA molecule mismatches out of six HLA molecules). In this sense banked CB-derived cells would be the future source for cell-based therapy. Moreover, these cells can be transduced with a different therapeutic gene to enhance angiogenesis or repair defective tissues.

Figure 2. Migration and accumulation of transgenic SPIO-labeled endothelial progenitor
cells in tumors. Ten million transgenic (hNIS-carrying) endothelial progenitor cells (EPCs; half of
them were labeled with SPIO) were intravenously administered in glioma-bearing rats after 14 days of
tumor implantation. MRI and technetium-99m SPECT were obtained on days 7 and 8, respectively.
(A) Preinjection MRI (before injection of labeled EPCs). (B) Postinjection MRI (after injection of
labeled EPCs). (C–E) Technetium-99m SPECT images in sagittal, coronal and axial plans. Note the low
signal intensity areas on MRI in tumor following intravenous administration of SPIO-labeled EPCs
(B, circle). SPECT images show increased accumulation of technetium-99m in the tumor compared
with the contralateral brain (white dotted areas, which showed almost no activity). (F) Prussian blue
and (G–I) immunohistochemistry stains show presence of iron-positive cells and expression of CD31
and hNIS.
FITC: Fluorescein isothiocyanate; NIS: Sodium iodide symporter; TRITC: Tetramethylrhodamine-5-(and
6)-isothiocyanate.
Adapted from [88].
Antiangiogenic treatments for malignant tumors are rapidly being developed. However, early reports indicate that these treatments often encounter development of tumor resistance to the treatment and activation of alternative pro-angiogenic pathways. EPCs, either genetically transduced or tagged with different contrast agents for different imaging modalities, can be used to determine the active process of neovascularization in these tumors. Like the determination of a hidden infection site using In-111-labeled white blood cells in nuclear medicine, In-111-labeled ECPs can be used to determine the status of neovascularization process in tumors or in lesions before and after anti- or pro-angiogenic treatments. In this case, EPCs would commonly be collected from patients PB after mobilization or from patients’ BM. However, having available stored CB EPCs that can be used to tag with contrast agents and used as imaging probes would be an extreme advantage.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
*of interest
* of considerable interest
- Risau W, Flamme I. Vasculogenesis. Ann. Rev. Cell. Dev. Biol. 11(1), 73–91 (1995).
- Folkman J, Shing Y. Angiogenesis. J. Biol. Chem. 267(16), 10931–10934 (1992).
- Zhang ZG, Zhang L, Jiang Q, Chopp M. Bone marrow-derived endothelial progenitor cells participate in cerebral neovascularization after focal cerebral ischemia in the adult mouse. Circ. Res. 90(3), 284–288 (2002).
- Asahara T, Murohara T, Sullivan A et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science 275(5302), 964–967 (1997). nn Pioneering work on endothelial progenitor cells.
- Gehling UM, Ergun S, Schumacher U et al. In vitro differentiation of endothelial cells from AC133-positive progenitor cells. Blood 95(10), 3106–3112 (2000).
- Janic B, Guo AM, Iskander AS, Varma NR, Scicli AG, Arbab AS. Human cord bloodderived AC133+ progenitor cells preserve endothelial progenitor characteristics after long term in vitro expansion. PLoS One 5(2), E9173 (2010). & Established optimal suspension cell culture conditions for long-term in vitro expansion of cord blood (CB) endothelial progenitor cells (EPCs).
- Mancuso P, Calleri A, Cassi C et al. Circulating endothelial cells as a novel marker of angiogenesis. Adv. Exp. Med. Biol. 522, 83–97 (2003).
- Arbab AS, Pandit SD, Anderson SA et al. Magnetic resonance imaging and confocal microscopy studies of magnetically labeled endothelial progenitor cells trafficking to sites of tumor angiogenesis. Stem Cells 24(3), 671–678 (2006). & Demonstrated capacity of MRI to track magnetically labeled CB EPCs.
- Ceradini DJ, Kulkarni AR, Callaghan MJ et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 10(8), 858–864 (2004).
- Arbab AS, Janic B, Knight RA et al. Detection of migration of locally implanted AC133+ stem cells by cellular magnetic resonance imaging with histological findings. FASEB J. 22(9), 3234–3246 (2008). & MRI tracking of locally implanted CB EPCs.
- Arbab AS, Frank JA. Cellular MRI and its role in stem cell therapy. Regen. Med. 3(2), 199–215 (2008).
- Khakoo AY, Finkel T. Endothelial progenitor cells. Ann. Rev. Med. 56, 79–101 (2005).
- Rafii S, Lyden D. Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat. Med. 9(6), 702–712 (2003).
- Ward MR, Stewart DJ, Kutryk MJ. Endothelial progenitor cell therapy for the treatment of coronary disease, acute MI, and pulmonary arterial hypertension: current perspectives. Catheter Cardiovasc. Interv. 70(7), 983–998 (2007).
- Sekiguchi H, Ii M, Losordo DW. The relative potency and safety of endothelial progenitor cells and unselected mononuclear cells for recovery from myocardial infarction and ischemia. J. Cell. Physiol. 219(2), 235–242 (2009).
- Tateishi-Yuyama E, Matsubara H, Murohara T et al. Therapeutic angiogenesis for patients with limb ischaemia by autologous transplantation of bone-marrow cells: a pilot study and a randomised controlled trial. Lancet 360(9331), 427–435 (2002).
- Jung KH, Roh JK. Circulating endothelial progenitor cells in cerebrovascular disease. J. Clin. Neurol. 4(4), 139–147 (2008).
- Sa Y, Hao J, Samineni D et al. Brain distribution and elimination of recombinant human TIMP-1 after cerebral ischemia and reperfusion in rats. Neurol. Res. 33(4), 433–438 (2011).
- Shi Q, Rafii S, Wu MH et al. Evidence for circulating bone marrow-derived endothelial cells. Blood 92(2), 362–367 (1998).
- Peichev M, Naiyer AJ, Pereira D et al. Expression of VEGFR-2 and AC133 by circulating human CD34+ cells identifies a population of functional endothelial precursors. Blood 95(3), 952–958 (2000).
- Handgretinger R, Gordon PR, Leimig T et al. Biology and plasticity of CD133+ hematopoietic stem cells. Ann. NY Acad. Sci. 996, 141–151 (2003).
- Miraglia S, Godfrey W, Yin AH et al. A novel five-transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood 90(12), 5013–5021 (1997).
- Ingram DA, Mead LE, Tanaka H et al. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood 104(9), 2752–2760 (2004).
- Nagano M, Yamashita T, Hamada H et al. Identification of functional endothelial progenitor cells suitable for the treatment of ischemic tissue using human umbilical cord blood. Blood 110(1), 151–160 (2007).
- Yin AH, Miraglia S, Zanjani ED et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood 90(12), 5002–5012 (1997).
- Fujiyama S, Amano K, Uehira K et al. Bone marrow monocyte lineage cells adhere on injured endothelium in a monocyte chemoattractant protein-1-dependent manner and accelerate reendothelialization as endothelial progenitor cells. Circ. Res. 93(10), 980–989 (2003).
- Duda DG, Cohen KS, Scadden DT, Jain RK. A protocol for phenotypic detection and enumeration of circulating endothelial cells and circulating progenitor cells in human blood. Nat. Protocols 2(4), 805–810 (2007).
- Zeoli A, Dentelli P, Rosso A et al. Interleukin-3 promotes expansion of hemopoietic-derived CD45+ angiogenic cells and their arterial commitment via STAT5 activation. Blood 112(2), 350–361 (2008).
- Kudo FA, Nishibe T, Nishibe M, Yasuda K. Autologous transplantation of peripheral blood endothelial progenitor cells (CD34+) for therapeutic angiogenesis in patients with critical limb ischemia. Int. Angiol. J. Int. Union Angiol. 22(4), 344–348 (2003).
- Bartunek J, Vanderheyden M, Vandekerckhove B et al. Intracoronary injection of CD133-positive enriched bone marrow progenitor cells promotes cardiac recovery after recent myocardial infarction: feasibility and safety. Circulation 112, I178–I183 (2005).
- Manginas A, Goussetis E, Koutelou M et al. Pilot study to evaluate the safety and feasibility of intracoronary CD133+ and CD133- CD34+ cell therapy in patients with nonviable anterior myocardial infarction. Catheter. Cardiovasc. Interv. 69(6), 773–781 (2007).
- Losordo DW, Schatz RA, White CJ et al. Intramyocardial transplantation of autologous CD34+ stem cells for intractable angina: a Phase I/IIa double-blind, randomized controlled trial. Circulation 115, 3165–3172 (2007).
- Ahmadi H, Baharvand H, Ashtiani SK et al. Safety analysis and improved cardiac function following local autologous transplantation of CD133(+) enriched bone marrow cells after myocardial infarction. Curr. Neurovasc. Res. 4(3), 153–160 (2007).
- Schots R, De Keulenaer G, Schoors D et al. Evidence that intracoronary-injected CD133+ peripheral blood progenitor cells home to the myocardium in chronic postinfarction heart failure. Exp. Hematol. 35(12), 1884–1890 (2007).
- Kawamoto A, Katayama M, Handa N et al. Intramuscular transplantation of G-CSFmobilized CD34(+) cells in patients with critical limb ischemia: a Phase I/IIa, multicenter, single-blinded, dose-escalation clinical trial. Stem Cells 27(11), 2857–2864 (2009).
- Scheubel RJ, Zorn H, Silber RE et al. Age-dependent depression in circulating endothelial progenitor cells in patients undergoing coronary artery bypass grafting. J. Am. Coll. Cardiol. 42(12), 2073–2080 (2003).
- Vasa M, Fichtlscherer S, Aicher A et al. Number and migratory capacity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ. Res. 89, 1–7 (2001).
- Hill JM, Zalos G, Halcox JP et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 348(7), 593–600 (2003).
- Murohara T, Ikeda H, Duan J et al. Transplanted cord blood-derived endothelial precursor cells augment postnatal neovascularization. J. Clin. Invest. 105(11), 1527–1536 (2000).
- Schlechta B, Wiedemann D, Kittinger C et al. Ex-vivo expanded umbilical cord blood stem cells retain capacity for myocardial regeneration. Circ. J. Off. J. Jap. Circ. Soc. 74(1), 188–194 (2010).
- Leor J, Guetta E, Feinberg MS et al. Human umbilical cord blood-derived CD133+ cells enhance function and repair of the infarcted myocardium. Stem Cells 24(3), 772–780 (2006).
- Ott I, Keller U, Knoedler M et al. Endothelial-like cells expanded from CD34+ blood cells improve left ventricular function after experimental myocardial infarction. FASEB J. 19(8), 992–994 (2005).
- Masuda H, Iwasaki H, Kawamoto A et al. Development of serum-free quality and quantity control culture of colony forming endothelial progenitor cell for vasculogenesis. Stem Cells Trans. Med. 1(2), (2012).
- Ahrens I, Domeij H, Topcic D et al. Successful in vitro expansion and differentiation of cord blood derived CD34+ cells into early endothelial progenitor cells reveals highly differential gene expression. PLoS One 6(8), E23210 (2011).
- O E, Lee BH, Ahn HY et al. Efficient nonadhesive ex vivo expansion of early endothelial progenitor cells derived from CD34+ human cord blood fraction for effective therapeutic vascularization. FASEB J. 25(1), 159–169 (2011).
- Stamm C, Westphal B, Kleine HD et al. Autologous bone-marrow stem-cell transplantation for myocardial regeneration. Lancet 361(9351), 45–46 (2003).
- Stamm C, Kleine HD, Westphal B et al. CABG and bone marrow stem cell transplantation after myocardial infarction. Thorac. Cardiovasc. Surg. 52(3), 152–158 (2004).
- Yossi C, Arnon N. Umbilical cord blood transplantation – how, when and for whom? Blood Rev. 18(3), 167–179 (2004).
- Mcguckin CP, Forraz N. Potential for access to embryionic-like cells from human umbilical cord blood. Cell Prolif. 41, 31–40 (2008).
- Yao CL, Feng YH, Lin XZ, Chu IM, Hsieh TB, Hwang SM. Characterization of serum-free ex vivo – expanded hematopoietic stem cells derived from human umbilical cord blood CD133+ cells. Stem Cells Dev. 15(1), 70–78 (2006).
- Weisel KC, Moore MaS, Kanz L, Möhle R. Extended in vitro expansion of adult, mobilized CD34+ cells without significant cell senescence using a stromal cell coculture system with single cytokine support. Stem Cells Dev. 18(2), 229–234 (2009).
- Gammaitoni L, Weisel KC, Gunetti M et al. Elevated telomerase activity and minimal telomere loss in cord blood long-term cultures with extensive stem cell replication. Blood 103(12), 4440–4448 (2004).
- Gluckman E. Ten years of cord blood transplantation: from bench to bedside. Br. J. Haematol. 147(2), 192–199 (2009). & Important review on the advantages of using CB as a source of progenitor cells for transplantation.
- Rocha V, Gluckman E. Improving outcomes of cord blood transplantation: HLA matching, cell dose and other graft- and transplantation-related factors. Br. J. Haematol. 147(2), 262–274 (2009).
- Rocha V, Wagner JE, Sobocinski KA et al. Graft-versus-host disease in children who have received a cord-blood or bone marrow transplant from an HLA-identical sibling. N. Engl. J. Med. 342(25), 1846–1854 (2000).
- Liu E, Law HK, Lau YL. Tolerance associated with cord blood transplantation may depend on the state of host dendritic cells. Br. J. Haematol. 126(4), 517–526 (2004).
- Madlambayan G, Rogers I. Umbilical cord-derived stem cells for tissue therapy: current and future uses. Regen. Med. 1(6), 777–787 (2006).
- Sacchi N, Costeas P, Hartwell L et al. Haematopoietic stem cell donor registries: World Marrow Donor Association recommendations for evaluation of donor health. Bone Marrow Trans. 42(1), 9–14 (2008).
- Suzanne MW, Marcela C. Stem cell medicine: umbilical cord blood and its stem cell potential. Semin. Fetal Neonatal Med. 10(3), 209–220 (2005).
- Yang C, Zhang ZH, Li ZJ, Yang RC, Qian GQ, Han ZC. Enhancement of neovascularization with cord blood CD133+ cell-derived endothelial progenitor cell transplantation. Thrombo. Haemosta. 91(6), 1202–1212 (2004).
- Hill JM, Finkel T, Quyyumi AA. Endothelial progenitor cells and endothelial dysfunction. Vox. Sang. 87(Suppl. 2), S31–S37 (2004).
- Senegaglia AC, Barboza LA, Dallagiovanna B et al. Are purified or expanded cord blood-derived CD133+ cells better at improving cardiac function? Exp. Biol. Med. 235(1), 119–129 (2010).
- Klein HM, Ghodsizad A, Marktanner R et al. Intramyocardial implantation of CD133+ stem cells improved cardiac function without bypass surgery. Heart Surg. Forum 10(1), E66–E69 (2007).
- Furfaro EM, Gaballa MA. Do adult stem cells ameliorate the damaged myocardium? Human cord blood as a potential source of stem cells. Curr. Vasc. Pharmacol. 5(1), 27–44 (2007).
- Chen J, Sanberg P, Li Y et al. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke J. Cerebral Circ. 32(11), 2682–2688 (2001).
- Nan Z, Grande A, Sanberg CD, Sanberg PR, Low WC. Infusion of human umbilical cord blood ameliorates neurologic deficits in rats with hemorrhagic brain injury. Ann. NY Acad. Sci. 1049, 84–96 (2005).
- Vendrame M, Gemma C, De Mesquita D et al. Anti-inflammatory effects of human cord blood cells in a rat model of stroke. Stem Cells Dev. 14(5), 595–604 (2005).
- Newcomb JD, Ajmo CT Jr, Sanberg CD, Sanberg PR, Pennypacker KR, Willing AE. Timing of cord blood treatment after experimental stroke determines therapeutic efficacy. Cell Trans. 15(3), 213–223 (2006).
- Xiao J, Nan Z, Motooka Y, Low WC. Transplantation of a novel cell line population of umbilical cord blood stem cells ameliorates neurological deficits associated with ischemic brain injury. Stem Cells Dev. 14(6), 722–733 (2005).
- Nystedt J, Mäkinen S, Laine J, Jolkkonen J. Human cord blood CD34+ cells and behavioral recovery following focal cerebral ischemia in rats. Acta Neurobiol. Exp. 66(4), 293–300 (2006).
- Boltze J, Reich DM, Hau S et al. Assessment of neuroprotective effects of human umbilical cord blood mononuclear cell subpopulations in vitro and in vivo. Cell Transplant. (2011).
- Willenbrock S, Knippenberg S, Meier M et al. In vivo MRI of intraspinally injected SPIO-labelled human CD34+ cells in a transgenic mouse model of ALS. In vivo 26(1), 31–38 (2012).
- Shi H, Yang W, Cui ZH et al. Tracking of CFSE-labeled endothelial progenitor cells in laser-injured mouse retina. Chin. Med. J. 124(5), 751–757 (2011).
- Zeng L, Ding S, Yan Z et al. Irradiation induces homing of donor endothelial progenitor cells in allogeneic hematopoietic stem cell transplantation. Int. J. Hematol. 95(2), 189–197 (2012).
- Grassinger J, Nilsson SK. Methods to analyze the homing efficiency and spatial distribution of hematopoietic stem and progenitor cells and their relationship to the bone marrow endosteum and vascular endothelium. Methods Mol. Biol. 750, 197–214 (2011).
- Takizawa H, Regoes RR, Boddupalli CS, Bonhoeffer S, Manz MG. Dynamic variation in cycling of hematopoietic stem cells in steady state and inflammation. J. Exp. Med. 208(2), 273–284 (2011).
- Burciaga-Nava JA, Reyes-Romero MA, Avelar-Gonzalez FJ, Guerrero-Barrera AL. Establishment and characterization of porcine aortic endothelial cell cultures with prolonged replicative lifespan by a non-enzymatic method. In Vitro Cell Dev. Biol. Anim. 45(1–2), 15–18 (2009).
- Duan HX, Lu GX, Cheng LM. Isolation, culture and identification of two types of endothelial progenitor cells from human umbilical cord blood. Zhongguo Shi Yan Xue Ye Xue Za Zhi 16(2), 387–391 (2008).
- Ma G, Lin XX, Jin YB et al. Isolation, culture and characterization of endothelial cells in infantile hemangioma. Zhonghua Zheng Xing Wai Ke Za Zhi 24(2), 144–147 (2008).
- Werner C, Bohm M, Friedrich EB. Role of integrin-linked kinase for functional capacity of endothelial progenitor cells in patients with stable coronary artery disease. Biochem. Biophys. Res. Commun. 377(2), 331–336 (2008).
- Che J, Doubrovin M, Serganova I et al. HSP70-inducible hNIS-IRES-eGFP reporter imaging: response to heat shock. Mol. Imaging 6(6), 404–416 (2007).
- Aboody-Guterman KS, Pechan PA, Rainov NG et al. Green fluorescent protein as a reporter for retrovirus and helper virus-free HSV-1 amplicon vector-mediated gene transfer into neural cells in culture and in vivo. Neuroreport 8(17), 3801–3808 (1997).
- Chen C, Zeng L, Ding S, Xu K. Adult endothelial progenitor cells retain hematopoiesis potential. Transplant. Proc. 42(9), 3745–3749 (2010).
- Laschke MW, Giebels C, Nickels RM, Scheuer C, Menger MD. Endothelial progenitor cells contribute to the vascularization of endometriotic lesions. Am. J. Pathol. 178(1), 442–450 (2011).
- Varma NR, Janic B, Ali MM, Iskander A, Arbab AS. Lentiviral based gene transduction and promoter studies in human hematopoietic stem cells (hHSCs). J. Stem Cells Regenerat. Med. 7(1), 41–53 (2011).
- Hayakawa J, Hsieh MM, Anderson DE et al. The assessment of human erythroid output in NOD/SCID mice reconstituted with human hematopoietic stem cells. Cell Trans. 19(11), 1465–1473 (2010).
- Swirski FK, Berger CR, Figueiredo JL et al. A near-infrared cell tracker reagent for multiscopic in vivo imaging and quantification of leukocyte immune responses. PLoS One 2(10), e1075 (2007).
- Varma NR, Janic B, Iskander AS et al. Endothelial progenitor cells (EPCs) as gene carrier system for rat model of human glioma. PLoS One 7(1), e30310 (2012).
- Chatterjee S, Lu D, Podsakoff G, Wong KK Jr. Strategies for efficient gene transfer into hematopoietic cells. The use of adenoassociated virus vectors in gene therapy. Ann. NY Acad. Sci. 770, 79–90 (1995).
- Chatterjee S, Wong KK Jr. Adeno-associated virus vectors for gene therapy of the hematopoietic system. Curr. Topics Microbiol. Immunol. 218, 61–73 (1996).
- Ma Y, Ramezani A, Lewis R, Hawley RG, Thomson JA. High-level sustained transgene expression in human embryonic stem cells using lentiviral vectors. Stem Cells 21(1), 111–117 (2003).
- Pfeifer A, Ikawa M, Dayn Y, Verma IM. Transgenesis by lentiviral vectors: lack of gene silencing in mammalian embryonic stem cells and preimplantation embryos. Proc. Natl Acad. Sci. USA 99(4), 2140–2145 (2002).
- Toelen J, Deroose CM, Gijsbers R et al. Fetal gene transfer with lentiviral vectors: long-term in vivo follow-up evaluation in a rat model. Am. J. Obstet. Gynecol. 196(4), 352. e1–352.e6 (2007).
- Dai G, Levy O, Carrasco N. Cloning and characterization of the thyroid iodide transporter. Nature 379(6564), 458–460 (1996).
- Smanik PA, Liu Q, Furminger TL et al. Cloning of the human sodium lodide symporter. Biochem. Biophys. Res. Commun. 226(2), 339–345 (1996).
- Barton KN, Xia X, Yan H et al. A quantitative method for measuring gene expression magnitude and volume delivered by gene therapy vectors. Mol. Ther. 9(4), 625–631 (2004).
- Chen L, Altman A, Mier W, Lu H, Zhu R, Haberkorn U. 99mTc-pertechnetate uptake in hepatoma cells due to tissue-specific human sodium iodide symporter gene expression. Nucl. Med. Biol. 33(4), 575–580 (2006).
- Rad AM, Iskander AS, Janic B, Knight RA, Arbab AS, Soltanian-Zadeh H. AC133+ progenitor cells as gene delivery vehicle and cellular probe in subcutaneous tumor models: a preliminary study. BMC Biotechnol. 9, 28 (2009).
- Higuchi T, Anton M, Saraste A et al. Reporter gene PET for monitoring survival of transplanted endothelial progenitor cells in the rat heart after pretreatment with VEGF and atorvastatin. J. Nucl. Med. 50(11), 1881–1886 (2009).
- Arbab AS. Activation of alternative pathways of angiogenesis and involvement of stem cells following anti-angiogenesis treatment in glioma. Histol. Histopathol. 27(5), 549–557 (2012).
- Arbab AS, Yocum GT, Rad AM et al. Labeling of cells with ferumoxides-protamine sulfate complexes does not inhibit function or differentiation capacity of hematopoietic or mesenchymal stem cells. NMR Biomed. 18(8), 553–559 (2005).
- Ju S, Teng G, Zhang Y, Ma M, Chen F, Ni Y. In vitro labeling and MRI of mesenchymal stem cells from human umbilical cord blood. Magn. Reson. Imaging 24(5), 611–617 (2006).
- Siow TY, Chen CC, Lin CY, Chen JY, Chang C. MR phase imaging: sensitive and contrast-enhancing visualization in cellular imaging. Magn. Reson. Imaging 30(2), 247–253 (2012).
- Hu SL, Lu PG, Zhang LJ et al. In vivo magnetic resonance imaging tracking of SPIO-labeled human umbilical cord mesenchymal stem cells. J. Cell Biochem. 113(3), 1005–1012 (2012).
- Islam T, Josephson L. Current state and future applications of active targeting in malignancies using superparamagnetic iron oxide nanoparticles. Cancer Biomark. 5(2), 99–107 (2009).
- Janic B, Rad AM, Jordan EK et al. Optimization and validation of FePro cell labeling method. PLoS One 4(6), E5873 (2009).
- Wang S, Fang J, Zhang T et al. Magnetic resonance imaging targeting of intracranial glioma xenografts by Resovist-labeled endothelial progenitor cells. J. Neurooncol. 105(1), 67–75 (2011).