Review Article - Imaging in Medicine (2010) Volume 2, Issue 3
Predicting the no-reflow phenomenon following successful percutaneous coronary intervention
L Galiuto†1, L Paraggio1, G Liuzzo1, AR de Caterina1and F Crea1
1Institute of Cardiology, Catholic University of the Sacred Heart, Policlinico A Gemelli, Largo A Gemelli, 8, 00168 Rome, Italy
- *Corresponding Author:
- L Galiuto
Institute of Cardiology, Catholic University of the Sacred Heart Policlinico A Gemelli
Largo A Gemelli, 8, 00168 Rome, Italy
Tel: +39 063 015 4187
Fax:+39 063 055 535
E-mail: lgaliuto@rm.unicatt.it
Abstract
In the setting of acute myocardial infarction, early and adequate reopening of an infarct-related artery is not necessarily followed by a complete restoration of myocardial perfusion. This condition is usually defined as ‘no-reflow’. The pathophysiology of no-reflow is multifactorial since extravascular compression, microvascular vasoconstriction, embolization during percutaneous coronary intervention, and platelet and neutrophil aggregates are involved. In the clinical arena, angiographic findings and easily available clinical parameters can predict the risk of no-reflow. More recently, several studies have demonstrated that biomarkers, especially those related to the pathogenetic components of no-reflow, could also have a prognostic role in the prediction and in the full understanding of the multiple mechanisms of this phenomenon. Thus, in this article, we investigate the role of several biomarkers on admission in predicting the occurrence of no-reflow after successful percutaneous coronary intervention.
Keywords
biomarkers; ; o-reflow; reperfusion injury; microvascular damage
The goal of primary percutaneous coronary intervention (PCI) in ST-segment elevation myocardial infarction (STEMI) is the rapid restoration of epicardial blood flow to minimize the progression of myocardial necrosis and to improve overall survival [1,2]. However, in almost 40% of patients, myocardial perfusion in the infarct-related artery territory has been shown to remain compromised despite the complete restoration of normal flow in the epicardial coronary artery. This condition has been defined as the ‘no-reflow’ phenomenon [3].
No-reflow phenomenon strongly affects the outcome of PCI, since patients with no-reflow phenomenon have been demonstrated to have a worse clinical prognosis than those in which myocardial perfusion is normally restored [4]. Recently, clinical research has focused on understanding, detection, prevention and treatment of this condition.
Once the pathophysiology, the assessment and the potential therapeutic approaches to noreflow is clarified, we would need a means of risk stratification in order to select patients at high risk of no-reflow.
In the setting of acute myocardial infarction (AMI), cardiac biomarkers, such as troponin T (cTnT), troponin I (cTnI) and creatin kinase (Ck)-MB, play an important role in the diagnosis and in the prognostic stratification of the patients, as underlined by guidelines of both the American College of Cardiology [5] and European Society of Cardiology [6]. However, only in recent years has the relationship between cTnT, cTnI and Ck-MB and the prevalence of no-reflow been investigated. More recently, several research groups have focused their attention on the role – in the setting of STEMI – of noncardiac biomarkers on admission, such as white blood cells (WBCs) and neutrophil count, mean platelet volume (MPV), C-reactive protein (CRP), hyperglycemia, fibrinogen and brain natriuretic peptide (BNP). The rising role of cardiac and noncardiac biomarkers in the prediction of no-reflow is strictly related to their possible involvement in distinct pathogenic mechanisms of this phenomenon.
The aim of the present article is a reappraisal of these biomarkers and of their possible usefulness in modifying the clinical approach and treatment of no-reflow phenomenon in patients with AMI.
Pathophysiology of no-reflow
Hystorically, the no-reflow phenomenon was first described in an ischemic canine heart by Kloner et al. [7]. In humans, the pathophysiology of no-reflow has been elucidated by several studies. Specifically, it has been reported that this phenomenon is mostly caused by the variable combination of four pathogenetic components: distal atherotrombotic embolization, ischemic injury, reperfusion injury and individual predisposition to microvascular damage (Figure 1).
Distal atherothrombotic embolization can originate from thrombus disruption during PCI [8]; indeed, microvascular blood flow decreases irreversibly when more than 50% of coronary capillaries are obstructed [9]. However, distal embolization seems to be a more complex phenomenon that involves several factors that are released from the site of lipid-rich plaque disruption. Liberation of plaque components, including platelet–fibrin complex, macrophages and colesterol crystals, might induce arteriolar spasm, leading to further microvascular congestion, thrombosis and reduced coronary perfusion.

Figure 1. Prinipal physiopathologic mechanisms involved in no-reflow phenomenon.
Ischemia is a major determinant of both microvascular and myocyte damage. However, microvessels are more resistant to ischemia than myocytes, as it has been demonstrated by the analysis of tissue damage after different ischemic periods. Accordingly, since the no-reflow area is always confined within the necrotic area, microvascular damage does not appear to be the primary cause of myocardial damage [10].
Reperfusion also carries undesirable effects and affects no-reflow. This phenomenon, commonly known as reperfusion injury, is mostly caused by the rapid initial influx of fluid and electrolytes that are responsible for the endothelium cell swelling and myocytes’ contraction band necrosis, followed by microvascular damage. In addition, the local rapid increase of oxygenated blood leads to the overproduction of oxygen free radicals and proinflammatory mediators by neutrophils (mostly in the first 2–10 min), which may increase endothelial dysfunction. This local increase of such proinflammatory mediators can also promote tissue edema and extravascular hemorrhage. Finally, platelets and neutrophils enhance the formation of leukocyte plugs and aggregates of red cells that have lost their flexibility [11,12]. Reperfusion might also cause irreversible damage to myocytes by triggering an uncontrolled hypercontraction, caused by calcium overload [13].
Finally, the individual predisposition to microvascular injury may be genetic or acquired and it has been mostly related to the presence of diabetes [14], hypercholesterolemia [15] and preconditioning [16,17]. Of note, a recent study suggests that the 1976T–C polymorphism of the adenosine 2A receptor is associated with an increased incidence of no-reflow phenomenon [18]. Furthermore, patients with no-reflow demonstrate a more compact fibrin network, possibly suggesting a genetic-mediated resistance to lysis [19].
Our group has recently demonstrated that no-reflow, as detected by myocardial contrast echocardiography (MCE) 24 h after successful PCI, improves over time in almost 50% of patients, thus, leading to a ‘new’ categorization of no-reflow as ‘reversible’ and ‘sustained’ [20]. Reversible no-reflow is probably caused by the functional microvascular changes that have been observed after a short (10–20 min) ischemic time in the animal model. Several factors may be involved, such as the decreased number of perfused capillaries, with a consequent reduction in absolute blood flow volume, the increased capillary permeability and, importantly, a condition of impaired vasodilatation. Interestingly, this vasomotor impairment is confined to microcirculation, since epicardial coronary arteries maintain their vascular reactivity after ischemia-reperfusion. Conversely, the sustained no-reflow pattern reflects the anatomical irreversible damage following a longer ischemic injury, which have been previously elucidated, and may represent myocardial necrosis [3].
Diagnosis
Over the years, different diagnostic approaches have been described to diagnose no-reflow and more so to predict this condition during PCI (Table 1).
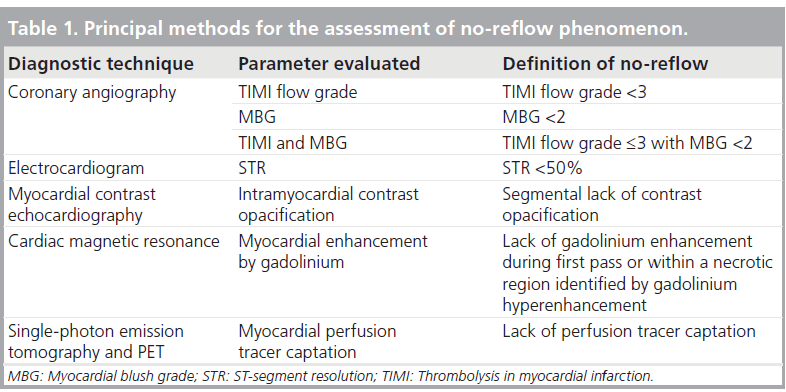
First of all, electrocardiographic ST-segment resolution (STR), as assessed 1 h after PCI, probably represents the most widely used technique, both in experimental studies and in clinical practice. Indeed, in the presence of impaired microvascular perfusion, ST-segment elevation persists despite successful PCI. The lack of STR over 50–70% is commonly considered diagnostic of no-reflow [21]. Sustained elevation of the ST segment after successful PCI is also associated with unfavorable functional and clinical outcomes. However, it has to be acknowledged that almost 30% of patients with thrombolysis in myocardial infarction (TIMI) flow 3 and myocardial blush grade (MBG) 2 or 3 do not exibit STR [22].
Several angiographic parameters can also be useful to detect no-reflow. First, the analysis of TIMI flow grade, which provides the rate of blood flow in the epicardial vessel, gives important information with regard to the occurrence of no-reflow [23], particularly TIMI flow grade scores from 0 (total absence of flow) to 3 (normal flow). TIMI flow grade 0–2, observed in 5–10% of patients after PCI, represents the angiographic evidence of no-reflow. The sensitivity of TIMI flow grade, however, is rather low as no-reflow occurs even in patients showing TIMI flow grade 3. MBG represents a newer and more sensitive method to assess no-reflow during PCI, since it provides a semi-quantitative evaluation of tissue perfusion after injection of contrast media in the epicardial vessel. Similarly to TIMI flow grade, MBG is scored on a scale of 0–3, with higher scores indicating better perfusion. An MBG of 0 or 1 is suggestive of no-reflow and has been observed in almost 50% of patients with TIMI flow grade 3 [24]. Among patients with TIMI flow grade 3, the assessment of MBG allows further risk stratification as only patients with normal epicardial flow and normal tissue perfusion have an extremely low risk of death. Accordingly, the diagnosis of no-reflow can be obtained by combining TIMI and MBG, defining no-reflow as TIMI flow grade 3 or less with MBG 0–1.
Coronary microvascular flow can be easily and noninvasively assessed by imaging techniques, thus, providing a direct visualization of myocardial perfusion and a quantification of myocardial blood flow. In the setting of STEMI, the most commonly used and validated techniques are cardiac magnetic resonance (CMR) and MCE.
Cardiac magnetic resonance uses gadolinium, a paramagnetic contrast media, to assess regional myocardial perfusion. Gadolinium is very useful in the evaluation of tissue perfusion as it behaves as an intravascular agent in the first phase after its administration. Using gadolinium-enhanced CMR, no-reflow can be diagnosed as: lack of gadolinium enhancement during first pass or lack of gadolinium enhancement within a necrotic region, identified by late gadolinium hyperenhancement [25]. CMR evaluation of microvascular perfusion has been shown to strictly correlate with MBG [26]. Finally, the detection of hypoenhancement zones on first-pass perfusion CMR, which represents no-reflow, is associated with permanent dysfunction at follow-up.
Myocardial contrast echocardiography uses ultrasounds to detect the presence of microbubbles in myocardial microvessels. Microbubbles are injected in the periferic circulation and easily bypass the pulmonary circulation reaching myocardial microvessels. The main component of microbubbles is air or high-molecular-weight gas, and their diameter and rheology are similar to that of erythrocytes. However, unlike red blood cells, they are able to scatter ultrasound waves and produce a signal detectable by echocardiographic probes. Microvascular obstruction is thus detectable as a perfusion defect during myocardial contrast echocardiography and represents the extent of no-reflow (Figure 2) [27,28]. Recently, in the AMI Contrast Imaging (AMICI) study, the extent of no-reflow has been demonstrated to be the best predictor of adverse left ventricular (LV) remodeling after STEMI, being superior to STR and MBG among patients with a TIMI flow grade 3 [29].

Figure 2. No-reflow as assessed by MCE. (A) Normal flow and (B) no-reflow area (arrows).
More recently, two new methods of assessing no-reflow have been carried out. A diastolic deceleration time (DDT) less than 600 ms 7 days after AMI, assessed by noninvasive transtoracic Doppler echocardiography, strictly predict LV dilation at 6 months. Therefore, DDT less than 600 ms 7 days after AMI was an independent predictor of LV remodeling and microvascular dysfunction [30]. Index of microcirculatory resistance is a new invasively assessed measure of microvasculature function using a pressure sensor/thermistor- tipped guidewire during PCI. Recently, the value of the index of microvascular resistance greater than 32 U (U = mean distal coronary pressure by the hyperemic transit time), assessed during primary PCI following STEMI, has been shown to strictly correlate with wall motion score at 3 months better than other angiographic measures of microvascular dysfunction [31].
In the clinical arena, angiographic parameters, such as TIMI flow grade and MBG, still represent the most useful tool to recognize noreflow. However, other techniques can help in clarifying the diagnosis and stratify the risk of these patients.
Other imaging methods used to assess microvascular patency, such as PET, myocardial scintigraphy and stress echocardiography, are difficult to use immediately after AMI and they are usually contraindicated in this clinical setting.
Clinical implications
Several studies have demonstrated that no-reflow has a negative impact on outcome, reducing the potential benefit of primary PCI. In fact, the occurrence of no-reflow is associated with a poor functional recovery and a higher incidence of early postinfarction complications, such as arrhythmias and development of congestive heart failure [32].
A large area of microvascular injury might impair the healing of the infarct area and could prevent the delivery of pharmacologic agents into that area. In fact, transmural damage is frequently associated with the no-reflow area; if the area of no-reflow is extended, infarct expansion and LV dilation are likely to occur. Therefore, no-reflow phenomenon is associated with a higher prevalence of adverse LV remodeling, which in turn, represents a strong predictor of late heart failure and mortality [29,33].
Since no-reflow phenomenon occurs in a reliable proportion of patients with AMI and its presence predicts a worse clinical outcome, the assessment of myocardial perfusion in the acute and subacute phase of AMI is useful for risk stratification in order to identify high-risk patients who need specific forms of treatment.
Therapeutic strategies
Several therapeutic strategies have been developed and tested to prevent and treat no-reflow. First, patients should undergo PCI as soon as possible since no-reflow occurrence and its severity are time dependent. Several trials have shown the importance of thrombus aspiration to prevent distal embolization after successful PCI, especially if a TIMI flow grade of no more than 1 is still present after guidewire insertion [34]. In fact, thrombus aspiration significantly reduces the extent of microvascular obstruction (assessed by MCE) and myocardial dysfunction. However, it does not have a significantly favorable effect in preventing LV remodeling [35] and could also have a possible deleterious effect, resulting in an increasing infarct size [36]. More recently, even the role of direct stenting [37] has been emphasized in this setting. Since platelet aggregation is one of the mechanisms involved in the no-reflow phenomenon, glycoprotein IIb/ IIIa receptor inhibitor, in the setting of STEMI, has been demonstrated to prevent no-reflow [38] and to reduce mortality and the rate of re-infarctions when administered during PCI.
Once the no-reflow phenomenon is established, other treatment options are available. Some pharmacologic agents, such as adenosine [39], nitroprusside [40] and nicorandil [41], seem to reduce the extension of microvascular damage after PCI as evaluated by electrocardiographic and angiographic indexes.
More recently, several new therapeutic approaches have been investigated. Sezer et al. have demonstrated that low-dose intracoronary administration of streptokinase immediately after PCI, improved myocardial reperfusion, although it does not improve long-term myocardial dysfunction [42]. On the other hand, Piot et al. have demonstrated a substantial reduction of infarct size after injection of cyclosporine at the time of reperfusion [43]. However, the beneficial effects of all these treatments as surrogate end points have not been associated with a better outcome.
Predictors of no-reflow
Clinical predictors
Several studies have evaluated the relationship between clinical, hemodynamic and electrocardiographic parameters obtained before PCI and incidence of no-reflow in patients with STEMI.
Cura et al. have demonstrated that the clinical characteristics of patients on admission, such as advanced age, elevated heart rate, angiographic evidence of thrombus and absence of coronary flow before PCI are independent predictors of TIMI flow grade 2 or less after PCI [44]. In the Assessment of Pexelizumab (APEX)-AMI trial, advanced age, patent artery and ischemic time demonstrated a strict association with the incidence of no-reflow after successful revascularization [45].
The duration of ischemia before PCI has been demonstrated to be the most important clinical predictor of no-reflow after successful PCI. Recently, Nallamothu et al. have demonstrated that a longer time to reperfusion is associated with a higher prevalence of no-reflow and with a larger no-reflow area [46]. Moreover, in a recent study by Iliceto et al., among 77 patients undergoing CMR after successful PCI, time-to-balloon was significantly associated at multivariate analysis both with myocardial transmural necrosis and severe microvascular obstruction [47].
Acute hyperglycemia occurs in up to 50% of all patients presenting with STEMI, regardless of a previous history of diabetes mellitus [48]. Irrespectively of the patient’s insulin sensitivity, acute hyperglycemia at admission is associated with worse clinical outcome and increased mortality in patients with AMI [49]. Several reasons may justify this increased risk: patients with hyperglycemia seem to present a larger infarct size, a higher incidence of congestive heart failure or cardiogenic shock and a tendency to arrhythmia [50]. Of note, an increased incidence of the no-reflow phenomenon has been associated with high admission glucose levels.
Several studies have been carried out to elucidate the role of acute hyperglycemia in the occurrence of no-reflow. Iwakura et al. have enrolled 146 consecutive patients with a first AMI and have assessed the occurrence of noreflow by myocardial contrast echocardiography after successful reperfusion [51]. They have demonstrated that, despite a similar proportion of diabetes mellitus and glycated hemoglobin values, patients with no-reflow after successful PCI, showed a significantly higher blood glucose level on admission than those with effective myocardial reperfusion (209 ± 79 mg/dl vs 159 ± 56 mg/dl, respectively; p < 0.0001). Moreover, at multivariate analysis, blood glucose level at admission was an independent predictor of no-reflow. More recently, Ishihara and colleagues have confirmed these findings prospectively demonstrating that, in 1253 consecutive patients with AMI, those patients with acute hyperglycemia (defined as plasma glucose level >198 mg/dl, regardless of the diabetic status) demonstrated a higher incidence of no-reflow (21 vs 12%; p < 0.001), while there was no difference in the incidence of no-reflow between diabetic and nondiabetic patients (14 vs 15%, respectively; p = 0.71) [52]. Neither of these studies clarify whether hyperglycemia was a cause or a consequence of a large infarct size that could be related to the no-reflow phenomenon.
Acute hyperglycemia also reduces the protective effect of preinfarction angina on microvascular function. In fact, Takahashy et al. found that, while in patients with normal glycemic levels at admission, preinfarction angina was associated with better angiographic parameters of microvascular function; in patients with acute hyperglycemia no significant difference with respect to these parameters were present between patients with and without preinfarction angina [53].
Finally, acute hyperglycemia at admission also influences the patient’s thrombotic state [54]. Indeed, elevated glucose levels at admission were associated with a significant increase in markers of thrombin formation, such as thrombin– antithrombin complexes (p = 0.0095) and prothrombin 1.2 fragments (p = 0.016), together with a higher level of CD40L (p < 0.0001), a marker of platelet activation and with reduced levels of fibrinogen (p = 0.002). Further prospective studies are needed to establish the possible benefit of tight glucose control before coronary reperfusion.
Angiographic predictors
Some angiographic parameters have been found to correlate with the prevalence of no-reflow. Above all, the involvement of the left anterior descending probably because of a larger extent of ischemic area [55].
A strict relationship between specific angiographic parameters and the prevalence of noreflow after successful PCI has been found by Yip et al. in 794 consecutive patients [56]; specifically, angiographic thrombus with the greatest linear dimension of more than three times the reference lumen diameter in the infarct-related artery, the presence of floating thrombus, a reference lumen diameter of the infarct-related artery that is larger than 4 mm, persistent contrast media distal to the obstruction and cutoff pattern (lesion morphology with an abrupt cutoff without taper before the occlusion) were found to correlate with a higher incidence of slow flow and no-reflow phenomenon after direct PCI.
Moreover, a recent study by Maekawa and colleagues has shown a major incidence of angiographic no-reflow in patients undergoing PCI with stent overexpansion (defined as stent:artery ratio of ≥1.2) [57].
Blood cell-related markers White blood cells & neutrophil count
It is well known that inflammation is a potent risk factor for the development of coronary artery disease. In recent years, the interest in the role of WBC and neutrophil count in the prediction of clinical outcome [58] and no-reflow in the setting of AMI has risen. The relationship between WBC count and the increased risk of AMI has long been recognized. More recent studies have highlighted the role of WBC count in the prediction of long and short-term outcome of patients with AMI [59,60].
The specific mechanism by which WBCs affect clinical outcome is, however, still under investigation. Barron et al. hypothesized that high WBC count could lead to increased morbidity and mortality, inducing a hypercoagulable or thromboresistant state [61]. It is well known that the major component of higher WBC count in the setting of AMI is represented by neutrophil count. Interestingly, in acute coronary syndromes, neutrophils are involved in plaque instability and several studies have demonstrated a general activation of these cells throughout the coronary tree [62]. Moreover, during reperfusion neutrophils can plug capillaries together with platelets [63] and they can release cytokines and neutrophil-derived mediators [64], such as oxygen free radicals and proteolytic enzymes, that reduce microvascular blood flow.
Kojima et al. have recently investigated the importance of admission WBC count as an independent predictor of no-reflow and mortality after successful PCI in the setting of AMI [65]. They retrospectively evaluated 1016 patients from the Japanese Coronary Syndrome Study database. They divided all patients in quartiles depending on WBC count and found significant differences between patients in highest quartile and patients in quartile one and two for the occurrence of no-reflow phenomenon, identified as TIMI flow grade of no more than 2 (p < 0.05). Patients in the highest quartile also had a hazard ratio of 2.9 compared with those in lowest quartile for death following AMI (p = 0.02). Moreover, WBC admission count resulted an independent predictor of no-reflow phenomenon following successful PCI (p = 0.025).
More recently, Sezer and colleagues prospectively demonstrated that, in 41 patients with AMI, a high neutrophil count at admission was strictly associated with the degree of microvascular damage, since there was a strong relationship between neutrophil count and angiographic analysis [66]. Moreover, they demonstrated a slightly significant relationship between admission WBC count and the same angiographic parameters. These associations were confirmed at multivariate analysis.
Recent investigations have suggested that myeloperoxidase, an enzyme secreted by activated neutrophils and monocytes, may be involved in the pathogenesis of coronary artery disease [67]. A recent study performed on 50 consecutive patients with AMI by Funayama et al. has demonstrated the strict association between myeloperoxidase and the no-reflow phenomenon, since the level of this enzyme was significantly greater at the culprit lesion in patients with no-reflow than those without no-reflow [68]. Moreover, they failed to demonstrate the difference in the plasma levels of elastase and IL-8 between the two groups of patients.
Therefore, WBC and neutrophil count on admission seem to be reliable markers of noreflow, reflecting the essential role of these cells in the pathophysiology of this phenomenon.
Mean platelet volume
Acute coronary syndromes are caused by an acute reduction of coronary f low, usually caused by the activation of an atherosclerotic plaque overlapped by a thrombus of variable extent, which may lead to coronary occlusion or subocclusion. In this setting, platelets play an important role. It has been demonstrated that MPV strictly correlates with their reactivity [69], since a greater MPV is associated with a higher expression of markers of platelet activity, such as glycoprotein Ib and glycoprotein IIb/IIIa receptors [70]. Several studies have also demonstrated that patients with unstable angina and AMI showed higher MPV when compared with patients with stable angina or noncardiac chest pain [71]. Moreover, MPV has been shown to be an independent risk factor for AMI [72] and strictly affect clinical outcome [73].
Platelets also play an important role in the occurrence of no-reflow phenomenon; the rush of platelet and neutrophils that follows reperfusion may lead to the formation of neutrophil–platelets aggregates that plug the microcirculation [63,74]. Huczek et al. have found that, in 398 consecutive patients presenting with STEMI, mean admission MPV was higher in patients with noreflow, assessed as TIMI flow grade less than 3, compared with those without no-reflow (10.8 vs 9.9 fl, respectively; p < 0.0001) [75]. Moreover, patients with high MPV, defined as a value in the third tertile (≥10.3 fl), had almost a fivefold higher risk of developing no-reflow as compared with to those with low MPV (21.2 vs 5.5%, respectively; p < 0.0001). At multivariate analysis, high MPV still remained a strong independent predictor of no-reflow (odds ratio [OR]: 4.7; p < 0.0001).
More importantly, MPV was found to be a predictor of no-reflow regardless of admission cTnI levels, since high MPV was strictly related with the occurrence of no-reflow in patients with both positive (p < 0.0001) and negative (p = 0.0027) admission cTnI levels. They also found that mean MPV value was significantly higher in those patients who died 6 months after AMI as compared with survivors (10.40 ± 0.85 vs 9.96 ± 0.90 fl, respectively; p = 0.0134). All these findings were confirmed by a more recent study by Sezer et al., in which MPV demonstrated a strict relation with angiographic parameters of reperfusion after successful PCI both at univariate and multivariate analysis, thus suggesting a future increasing role of MPV in clinical arena [66].
Tromboxane A2
Niccoli et al. have recently aimed to assess the role of plasma levels of tromboxane A2 (TXA2) at admission in predicting the occurrence of noreflow phenomenon [76]. It is well known that TXA2 is a key mediator of platelet activation and aggregation, and an important mediator of platelet-induced coronary vasoconstriction. They have, thus, supposed that TXA2 might be involved in platelet activation and plugging, both occurring in no-reflow. A total of 47 consecutive patients with first STEMI undergoing PCI within 12 h of onset of symptoms were enrolled. The study demonstrated that higher admission plasma levels of TXA2 are associated with higher incidence of microvascular injury following successful PCI (17.74 vs 3.91 pg/ml; p = 0.005). Moreover, TXA2 admission plasma levels were also higher in patients lacking STR following PCI (19.58 vs 3.99 pg/ml; p = 0.001). Finally, multivariate analysis demonstrated that admission plasma levels of TXA2 were predictors of both angiographic no-reflow (OR: 1.08; p = 0.04) and lack of STR (OR: 1.13; p = 0.013).
These findings could suggest new therapeutic approaches to specifically block TXA2. Interestingly terutroban, a specific thromboxane receptor antagonist, has already been found to be more effective than aspirin in the inhibition of thrombus formation in animals [77].
Markers of myocardial necrosis Creatinkinase
Until the late-1980s the MB isoform of Ck represented the only specific marker of myocardial cells and the only biomarker that allows an easy assessment of myocardial necrosis. However, Ck-MB doesn’t have a high diagnostic specificity. Therefore, in the diagnosis of AMI, Ck/ Ck-MB are used only when cTnT and cTnI assays are not available. Moreover, several studies have demonstrated that both admission and peak Ck/Ck-MB values provide a lower prognostic value than cardiac troponins in the setting of AMI [78,79]. However, Ck/Ck-MB represents cytoplasmatic enzymes of myocardiocytes and they are rapidly released in systemic circulation in response to myocardial damage. For this reason, Ck/Ck-MB are, nowadays, the most effective biomarkers in providing an early diagnosis of AMI (within first 6 h).
Only a few studies have investigated the relationship between Ck/Ck-MB admission levels and the occurrence of no-reflow phenomenon. Only one study has demonstrated that Ck admission levels showed significant correlation with CMR no-reflow, assessed as delayed enhancement score of no more than 1 (p = 0.018) [80], while two other studies have demonstrated a correlation between no-reflow, assessed by TIMI flow grade less than 3 and Ck-MB peak value [56,81]. However, in no case were Ck/Ck-MB levels investigated in a multivariate analysis and many studies did not demonstrate significant correlation between these biomarkers and the occurrence of no-reflow, so the role of this biomarker in the prediction of no-reflow has to be considered still under investigation.
Cardiac troponin
Clinical outcome of patients with AMI is strongly affected by the occurrence and the extent of myocardial necrosis. cTnT and cTnI represent the tropomyosine binding protein of the contractile apparatus of cardiac myocytes. A direct correlation has been found between the extent of necrotic area (commonly known as ‘infarct size’) and both peak and area under the curve of cTnT [82]. Therefore, cTnT can be considered a sensitive and specific marker of myocardial necrosis. Several studies have demonstrated a strict relationship between positive admission levels of cTnT and major cardiac events and death [83,84], also among patients with TIMI flow grade 3, as shown by Kurowski et al. [85]. This association is probably due to major extent of necrotic area, less myocardial salvage and probably microvascular dysfunction.
Although many studies have investigated the relationship between cTnT and cTnI levels and clinical outcome, and between these biomarkers and infarct size, only few studies have focused on the role of cTnT and cTnI in predicting the occurrence of no-reflow phenomenon in the setting of STEMI. In the TIMI 10B study, admission levels of cTnI were significantly lower in the patients who demonstrated a TIMI flow grade 3 at 60-min angiogram after thrombolytic therapy [86], while in another study, these levels were lower in patients with TIMI flow grade 3, 90 min after trombolysis [87]. Matetzky and colleagues have demonstrated for the first time that 24% of 110 consecutive patients with elevated admission cTnI levels failed to achieve a TIMI flow grade 3, while patients with cTnI less than 0.4 ng/ml achieved this value in all cases [88].
A more recent study, carried out on 140 consecutive patients admitted with the diagnosis of first STEMI by Giannistis et al., has clarified the role of cTnT for the prediction of no-reflow [89]. They demonstrated that epicardial flow remained more frequently compromised (TIMI flow grade <3) in patients with a cTnT-positive value on admission than in cTnT-negative patients (25 vs 9%, respectively; p = 0.009). At multivariate analysis, cTnT was the most powerful predictor of successful PCI (TIMI flow grade 3), even superior to time to balloon.
Several other studies investigating the impact of clinical, angiographic and laboratoristic parameters for the prediction of no-reflow, have investigated the role of cTnT or cTnI. Huczek et al. found that admission-positive cTnI was an independent predictor of no-reflow (OR: 4.8; p = 0.004) [63], while Hong et al. have demonstrated that admission cTnT level was significantly higher in the no-reflow group than in normal flow group (p = 0.001) [90]. More recently, a study carried out in 27 patients with AMI by Porto et al. demonstrated that elevated admission cTnT was associated with perfusion defect size at first-pass CMR (n = 27, r = 0.42; p = 0.028) [91].
The role of biomarkers of myocardial necrosis in the prediction of no-reflow should, however, be clarified as they seem to be more strictly related with infarct size and, as a consequence, to myocardial cell necrosis more than microvascular damage.
Markers of inflammation & endothelial activation C-reactive protein
Systemic inflammation has been demonstrated to play an important role in all phases of atherosclerosis [92]. In addition, inf lammation enhances epicardial and microvascular tone [93]. Of note, myocardial injury that occurs during AMI generates an acute-phase reaction, as is the case for any kind of tissue injury [94], and the degree of this reaction may influence the clinical outcome of these patients [95]. Moreover, there has been an increasing interest in acute-phase reactant CRP in the prediction of cardiovascular events. In AMI, CRP has been localized in the infarcted region together with activated fragments of the complement system, where it may play a pathogenetic role by enhancing local inflammation [96]. Admission CRP levels have been demonstrated to affect final infarct size, probably owing to the detrimental effect of CRP on myocardium through complementmediated tissue damage [97]. Several recent studies have also demonstrated that high baseline CRP levels in patients with AMI undergoing primary PCI could predict early complications and worse short-term prognosis [98].
In the setting of AMI, activation of inflammatory cells may enhance no-reflow. Specifically, inflammation may contribute to microvascular obstruction through several mechanisms [95,99]: endothelial activation, impairment of microvascular response to both endothelium-dependent and endothelium-independent vasodilator stimuli and reduction of fibrinolytic response following vessel occlusion.
In a recent published article, Celik et al. demonstrated that, in 75 consecutive patients in the setting of AMI, admission CRP levels of patients with TIMI perfusion grade (an analog of MBG) 0–1 after successful PCI were significantly higher than those of patients with TIMI perfusion grade 2–3 (27.67 ± 8.31 vs 12.03 ± 3.95 μg/dl, respectively; p < 0.001) [100]. Moreover, CRP levels demonstrated a significant independent correlation with the development of poor myocardial perfusion after successful PCI (p = 0.003). More recently, Jesel et al. have demonstrated that CRP levels were also strictly associated with microvascular obstruction evaluated by CMR (n = 50, r = 0.43, respectively; p = 0.003) [67].
However, a recent study carried out by Niccoli et al. on 60 consecutive patients with first STEMI have shown no correlation between CRP admission levels in patients with final TIMI flow grade no more than 2 and those in patients with TIMI flow grade 3 (6 vs 3 mg/l; p = 0.1) [101]. Moreover, CRP admission levels were also similar between patients with MBG 2 or less, compared with those with MBG 3 (3 vs 3.8 mg/l, respectively; p = 0.8).
C-reactive protein-mediated complement activation together with neutrophil plugging may be the factors contributing to the development of microvascular damage in patients with AMI. Thus, the impact of CRP admission levels and those of complement system on predicting the occurrence of no-reflow has not been clearly elucidated and more studies are needed.
Fibrinogen
After successful PCI following an AMI, the alterations of blood rheology are more pronounced in patients with incomplete myocardial microcirculation reperfusion [102]. These patients mainly show enhanced erythrocyte aggregation and higher plasma viscosity, which elevates blood flow resistance in the microvasculature, thus, contributing to the occurrence of no-ref low phenomenon [103]. Fibrinogen concentration at admission is associated with adverse clinical outcome in patients with unstable angina and strictly correlates with infarct size [104], as assessed by Ck-MB peak value, and extent of no-perfusion areas at thallium scintigraphy.
Recently, Wasilewski et al. demonstrated that, in 105 consecutive patients with STEMI, high fibrinogen levels are associated with the absence of complete myocardial reperfusion, assessed by STR less than 50%, after successful PCI, as compared with patients with complete reperfusion (523 ± 198.02 vs 395.56 ± 144.98 mg/dl, respectively; p = 0.0004) [105]. Both at uni- and multivariate analysis, fibrinogen concentration has been found to be an independent predictor of noreflow phenomenon (OR: 1.56; p = 0.0023 and OR: 1.51; p = 0.021, respectively). These data have been confirmed by Hong et al., showing that patients who develop no-reflow had higher levels of fibrinogen concentration at admission, as compared with those with normal reflow (347 ± 112 vs 278 ± 83 mg/dl, respectively; p = 0.035) [90].
These findings may indicate an indirect role of high fibrinogen concentration, as it could lead to red blood cell aggregation in the occurrence of no-reflow phenomenon, which probably presents itself as an elevation of blood-flow resistance.
Von Willebrand factor
More recently, Sgueglia and colleagues demonstrated that plasma admission levels (reported as a percentage of normality) of von Willebrand Factor (vWF), which is known to play an important role in endothelial activation, were higher in patients with no-reflow than in those with optimal myocardial perfusion (160 ± 62 vs 121 ± 49%, respectively; p = 0.012, n = 54) [106]. Furthermore, a vWF plasma level of at least 150% was a significant predictor of no-reflow at multivariate analysis (OR: 3.8; p = 0.033). Accordingly, the most important clinical implication of this study is to improve the treatment of no-ref low by targeting the platelet–vWF interaction.
Endothelin
Since its initial discovery in 1988 by Yanagisawa et al. [107], the physiological and pathophysiological roles of endothelin (ET) have been extensively investigated. ET-1 is the most abundant of the ET peptides in the cardiovascular system and it is produced by endothelial cells, myocardiocytes and vascular smooth muscle cells [108]. ET-1 is a potent endothelium-derived vasoconstrictor peptide [109], an important modulator of neutrophil function [110] and a stimulator of surface expression of adhesion molecules [111]. ET-1 has been found to be elevated in the first hours following an AMI [112] and is a strong predictor of adverse outcome in patients with AMI following successful PCI.
The efficacy of a variety of ET receptor antagonists in the treatment of ischemia-reperfusion injury has been tested in different animal models, and conflicting results have been reported. However, in several of these studies, ET antagonists were administered before the onset of ischemia and continuously during reperfusion [113,114]. Although such administration was effective in most cases, it is not clinically applicable. More recently Galiuto et al. demonstrated that, in the canine model, the intravenous administration of an ET-1 antagonist immediately after reperfusion (a timing of administration potentially applicable in patients with AMI undergoing PCI) of 90 min of coronary occlusion preserved microvascular flow during reperfusion and prevented the increase in myocardial wall [115].
A recent study, carried out by the same group, which assessed the predictive role of TXA2 and vWF, has shown in 51 consecutive patients with STEMI, the association between admission plasma levels of ET-1 and microvascular injury in humans [116]. They found that higher ET-1 levels at admission were associated with the occurrence of no-reflow, defined as TIMI flow grade less than 3 and/or TIMI flow grade 3 with MBG no more than 2 (3.95 ± 0.7 vs 3.30 ± 0.8 pg/ml, respectively; p = 0.004). Moreover, at multivariate logistic analysis, ET-1 was an independent predictor of no-reflow (OR: 2.76; p = 0.03). However, no clinical outcome assessment has been performed as the population was too small. Similar findings have been carried out by a later study from the same group.
Moreover, Adlbrecht et al. demonstrated the role of thrombus-bound ET in determining microvascular dysfunction [117]. Specifically, they convincingly demonstrated, in a porcine cardiac model, that coronary vasoconstriction was enhanced by the injection of human coronary thrombus homogenates obtained from culprit vessels in patients undergoing PCI after AMI.
Finally, these data may have relevant clinical implications and a randomized trial on the effects of an ET-1 antagonist on no-reflow in humans is needed.
Tissue factor
The contribution of the coagulation system to the pathogenesis of no-reflow phenomenon has only recently been emphasized. Tissue factor (TF) is known to initiate blood coagulation by binding activated coagulation factor VII [118], thus, creating a complex that proteolytically activates factors IX and X, and triggers the coagulation system activation [119]. Moreover, TF is particularly abundant in coronary atherosclerotic plaques [120]. Therefore, the spontaneous mechanical disruption of atherosclerotic plaques might induce the release of active TF into coronary blood.
Bonderman et al. recently found, in 11 consecutive patients with first AMI, that TF antigen levels were more elevated in patients with no-reflow compared with control subjects, and even more elevated than in those with normal flow after successful PCI [121]. Moreover, in a pig model, they demonstrated the pathogenic role of TF in no-reflow phenomenon by injecting human TF containing atherosclerotic plaque material into porcine coronary circulation and detecting no-reflow in nine out of ten pigs within 30 s.
However, TF, as with many other biomarkers investigated in this article, shows very limited clinical application owing to the absence of a simple and quick diagnostic method. Furthermore, several clinical studies are needed to investigate whether anti-TF agents could offer new treatment opportunities.
Brain natriuretic peptide
Brain natriuretic peptide is a neurohormone that is synthesized and released by cardiac ventricles in response to an increased wall tension and direct myocardial damage [122]. BNP is produced as pro-hormone (pro-BNP), that is then cleaved into BNP and N-terminal pro-BNP (NT-pro-BNP). In the setting of AMI, BNP and NT-pro-BNP levels in the subacute phase are potent predictors of both short- and longterm mortality [123]. Goetze et al. demonstrated that myocardial ischemia is associated with an increase of both BNP and NT-pro-BNP values, even in the absence of left ventricular dysfunction [124]. In clinical practice, these biomarkers could represent a valuable screening test for cardiac disease in patients with a normal electrocardiogram, echocardiogram and cardiac enzymes [90].
In recent years, the possible correlation between BNP admission levels and the occurrence of no-reflow phenomenon has been evaluated. Grabowski et al. first demonstrated that, in 126 consecutive patients undergoing PCI after STEMI, baseline BNP levels were higher among patients with a final TIMI flow grade 2 or less after successful PCI (356.7 ± 350.8 vs 142.8 ± 191.2 pg/ml; p < 0.0001) and among patients with no-ref low, defined as TIMI flow grade less than 3 and no STR after PCI (427.3 ± 362.9 vs 142.8 ± 188.3 pg/ml; p < 0.0001) [125]. Furthermore, at multivariate analysis, a serum baseline BNP level over 100 pg/ml was an independent predictor of TIMI flow grade 2 or less and no-reflow, and the risk of developing no-reflow was almost fivetimes higher than in patients with BNP levels less than 100 pg/ml. Recently, Jeong and colleagues confirmed these findings, demonstrating that a BNP admission level over 90 pg/ml was an independent predictor of no-reflow (n = 300, OR: 14.953; p = 0.001) [126].
Hong et al. demonstrated that also NT-pro-BNP admission levels collected in 159 consecutive patients with STEMI are a reliable predictor of no-ref low phenomenon [90]. In fact, they demonstrated that baseline NT-pro-BNP levels were significantly higher in patients with no-reflow than in those without (1982 ± 3314 vs 415 ± 632 pg/ml, respectively; p = 0.005). At multivariate analysis, serum admission NT-pro-BNP levels over 500 pg/ ml were associated with a higher probability of developing no-reflow (OR: 4.42; p = 0.028).
However, all these studies failed to elucidate the underlying mechanism to the link between BNP and the no-reflow phenomenon, and more clinical and sperimental studies are needed to assess whether no-reflow can be the cause or the consequence of higher BNP.
Future perspective
The increasing understanding of the molecular mechanisms involved in no-reflow phenomenon after successful PCI have led to the evaluation of several new biomarkers as hypothetical predictors of no-reflow occurrence.
Most of the biomarkers reviewed in this article have been shown to be strictly related to the incidence of no-reflow phenomenon (Table 2). Of note, admission levels of several of these biomarkers, such as WBC and neutrophil count, CRP and MPV can be easily assessed in clinical practice. cTnI and cTnT have demonstrated the ability to predict the occurrence of no-reflow; however, they seem to be more strictly related to necrotic damage than microvascular damage. Therefore, in the setting of AMI, all these biomarkers could already play an important role in the risk stratification of patients showing no-reflow after successful PCI. However, it should be noticed that the levels of these biomarkers in systemic circulation do not necessarily mirror their concentration at the microvascular level, where no-reflow phenomenon occurs.

Several more biomarkers, such as chromogranin A and those related with complement pathway, apoptosis and T-cell activation, have been demonstrated to be strictly related to ischemia-reperfusion injury in animal models. However, their role has not been demonstrated in human tissue. It is of note that there is a lack of a quick, simply reproducible and accurate marker to assess the occurrence of apoptosis in clinical practice.
Several therapeutic strategies against noref low have been tested with inconsistent results, mostly because they have been applied to the totality of patients without considering that the relevance of each pathogenetic component of no-reflow differ from patient to patient. Blood elevation of specific biomarkers might reflect the prevalence of a specific pathogenetic mechanism, thus, leading to a personalized treatment aimed at counteracting the prevalent mechanisms of no-reflow in the specific patient. However, so far, there is still a lack of evidence to support the use of threshold values of a specific biomarker as inclusion criteria of a personalized treatment strategy.
However, more future studies on predicting roles of newer biomarkers investigated only in animal settings and involved in molecular mechanisms of no-reflow phenomenon should be carried out and several prospective studies are needed in order to shed new light with regard to the importance of each biomarkers, together with its corresponding molecular mechanism, in the occurrence of no-reflow in the single patient.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.

No writing assistance was utilized in the production of this manuscript.
Papers of special note have been highlighted as:
* of considerable interest
References
- Goldberg RJ, Glatfelter K, Burbank-Schmidt E, Lessard D, Gore JM: Trends in community mortality due to coronary heart disease. Am. Heart J. 151, 501–507 (2006).
- Armstrong PW, Granger CB, Adams PX et al.: Pexelizumab for acute ST-elevation myocardial infarction in patients undergoing primary percutaneous coronary intervention: a randomized controlled trial. JAMA 297, 43–51 (2007).
- Galiuto L, DeMaria AN, Iliceto S: Microvascular damage during myocardial ischemia-reperfusion: pathophysiology, clinical implications and potential therapeutic approach evaluated by myocardial contrast echocardiography. Ital. Heart J. 1(2), 108–116 (2000). & Very exhaustive paper regarding clinical implication and first therapeutical approaches to no-reflow, assessed by myocardial contrast echocardiography.
- Ito H, Maruyama A, Iwakura K et al.: Clinical implications of the no-reflow phenomenon. A predictor of complications and left ventricular remodelling in reperfused anterior wall myocardial infarction. Circulation 93, 223–228 (1996).
- Kushner FG, Hand M, Smith SC Jr et al.; American College of Cardiology Foundation/ American Heart Association Task Force on Practice Guidelines: 2009 Focused Updates: ACC/AHA Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction (Updating the 2004 Guideline and 2007 Focused Update) and ACC/AHA/SCAI Guidelines on Percutaneous Coronary Intervention (Updating the 2005 Guideline and 2007 Focused Update). Circulation 120, 2271–2306 (2009).
- Van de Werf F, Bax J, Betriu A et al.: Management of acute myocardial infarction in patients presenting with persistent ST-segment elevation. Eur. Heart J. 29(3), 2909–2945 (2008).
- Kloner RA, Ganote CE, Jennings RB: The “no-reflow” phenomenon after temporary coronary occlusion in the dog. J. Clin. Invest. 54, 1496–1508 (1974). & Historical paper in which no-reflow phenomenon has been described for the first time in a canine model after experimental occlusion of epicardial coronary artery.
- Skyschally A, Leineweber K, Gres P, Haude M, Erbel R, Heusch G: Coronary microembolization. Basic Res. Cardiol. 101, 373–382 (2006).
- Hori M, Inoue M, Kitakaze M, Koretsune Y et al.: Role of adenosine in hyperemic response of coronary blood flow in microembolization. Am. J. Physiol. 250, H509–H518 (1986).
- Ambrosio G, Weisman HF, Mannisi JA, Becker LC: Progressive impairment of regional myocardial perfusion after initial restoration of postischemic blood flow. Circulation 80, 1846–1861 (1989).
- Engler RL, Schmid-Schönbein GW, Pavelec RS: Leukocyte capillary plugging in myocardial ischemia and reperfusion in the dog. Am. J. Pathol. 111, 98–111 (1983).
- Ambrosio G, Tritto I: Reperfusion injury: experimental evidence and clinical implications. Am. Heart J. 138, S69–S75 (1999).
- Nakamura TY, Goda K, Okamoto T et al.: Contractile and morphological impairment of cultured fetal mouse myocytes induced by oxygen radicals and oxidants. Correlation with intracellular Ca2+ concentration. Circ. Res. 73(4), 758–770 (1993).
- Collet JP, Montalescot G: The acute reperfusion management of STEMI in patients with impaired glucose tolerance and Type 2 diabetes. Diabetes Vasc. Dis. Res. 2, 136–143 (2005).
- Golino P, Maroko PR, Carew TE: The effect of acute hypercholesterolemia on myocardial infarct size and the no-reflow phenomenon during coronary occlusion-reperfusion. Circulation 75, 292–298 (1987).
- Colonna P, Cadeddu C, Montisci R et al.: Reduced microvascular and myocardial damage in patients with acute myocardial infarction and preinfarction angina. Am. Heart J. 144(5), 796–803 (2002).
- Karila-Cohen D, Czitrom D, Brochet E et al.: Decreased no-reflow in patients with anterior myocardial infarction and pre-infarction angina. Eur. Heart J. 20, 1724–1730 (1999).
- Vignali L, Talanas G, Saia F et al.: Genetic association between the 1976T_C polymorphism in the adenosine A2 receptor and angiographic no-reflow phenomenon. Il Giornale Italiano di Cardiologia Invasiva 3(Suppl. 1), 109 (2007).
- Zalewski J, Undas A, Godlewski J, Stepien E, Zmudka K: No-reflow phenomenon after acute myocardial infarction is associated with reduced clot permeability and susceptibility to lysis. Arterioscler. Thromb. Vasc. Biol. 27, 2258–2265 (2007).
- Galiuto L, Lombardo A, Maseri A et al.: Temporal evolution and functional outcome of no-reflow: sustained and spontaneously reversible patterns following successful coronary recanalization. Heart 89, 731–737 (2003). & Pioneering paper that demonstrated, for the first time, the occurrence of two different patterns of no-reflow: sustained and reversible.
- Schröder R: Prognostic impact of early ST-segment resolution in acute ST-elevation myocardial infarction. Circulation 110, e506–e510 (2004).
- Giugliano RP, Sabatine MS, Gibson CM et al.: Combined assessment of thrombolysis in myocardial infarction flow grade, myocardial perfusion grade, and ST-segment resolution to evaluate epicardial and myocardial reperfusion. Am. J. Cardiol. 93, 1362–1367 (2004).
- The TIMI Study Group: The Thrombolysis In Myocardial Infarction [TIMI] trial. Phase I findings. N. Engl. J. Med. 312, 932–936 (1985).
- van’t Hof AW, Liem A, Suryapranata H, Hoorntje JC, de Boer MJ, Zijlstra F; Zwolle Myocardial Infarction Study Group: Angiographic assessment of myocardial reperfusion in patients treated with primary angioplasty for acute myocardial infarction: myocardial blush grade. Circulation 97, 2302–2306 (1998).
- Albert TS, Kim RJ, Judd RM: Assessment of no-reflow regions using cardiac MRI. Basic Res. Cardiol. 101, 383–390 (2006).
- Porto I, Burzotta F, Brancati M et al.: Relation of myocardial blush grade to microvascular perfusion and myocardial infarct size after primary or rescue percutaneous coronary intervention. Am. J. Cardiol. 99, 1671–1673 (2007).
- Iliceto S, Marangelli V, Marchese A, Amico A, Galiuto L, Rizzon P: Myocardial contrast echocardiography in acute myocardial infarction. Pathophysiological background and clinical applications. Eur. Heart J. 17, 344–353 (1996).
- Hayat SA, Senior R: Myocardial contrast echocardiography in ST elevation myocardial infarction: ready for prime time? Eur. Heart J. 29, 299–314 (2008).
- Galiuto L, Fedele F, Agati L; AMICI Investigators: The extent of microvascular damage during myocardial contrast echocardiography is superior to other known indexes of post-infarct reperfusion in predicting left ventricular remodeling: results of the multicenter AMICI study. J. Am. Coll. Cardiol. 51(5), 552–559 (2008).
- Shintani Y, Ito H, Iwakura K et al.: Usefulness of impairment of coronary microcirculation in predicting left ventricular dilation after acute myocardial infarction. Am. J. Cardiol. 93(8), 974–978 (2004).
- Fearon WF, Shah M, Ng M et al.: Predictive value of the index of microcirculatory resistance in patients with ST-segment elevation myocardial infarction. J. Am. Coll. Cardiol. 51(5), 560–565 (2008).
- Ito H, Tomooka T, Sakai N et al.: Lack of myocardial perfusion immediately after successful thrombolysis. A predictor of poor recovery of left ventricular function in anterior myocardial infarction. Circulation 85, 1699–1705 (1992).
- Bolognese L, Carrabba N, Parodi G et al.: Impact of microvascular dysfunction on left ventricular remodeling and long-term clinical outcome after primary coronary angioplasty for acute myocardial infarction. Circulation 109, 1121–1126 (2004).
- Burzotta F, Trani C, Romagnoli E et al.: Manual thrombusaspiration improves myocardial reperfusion: the randomized evaluation of the effect of mechanical reduction of distal embolization by thrombus-aspiration in primary and rescue angioplasty (REMEDIA) trial. J. Am. Coll. Cardiol. 46, 371–376 (2005).
- Galiuto L, Garramone B, Burzotta F et al.; REMEDIA Investigators: Thrombus aspiration reduces microvascular obstructionafter primary coronary intervention: a myocardial contrast echocardiographysubstudy of the REMEDIA Trial. J. Am. Coll. Cardiol. 48(7), 1355–1360 (2006).
- Kaltoft A, Bøttcher M, Nielsen SS et al.: Routine thrombectomy in percutaneous coronary intervention for acute ST-segmentelevation myocardial infarction: a randomized, controlled trial. Circulation 114(1), 40–47 (2006).
- Loubeyre C, Morice MC, Lefèvre T, Piéchaud JF, Louvard Y, Dumas P: A randomized comparison of direct stenting with conventional stent implantation in selected patients with acute myocardial infarction. J. Am. Coll. Cardiol. 39, 15–21 (2002).
- Thiele H, Schindler K, Friedenberger J et al.: Intracoronary compared with intravenous bolus abciximab application in patients with ST-elevation myocardial infarction undergoing primary percutaneous coronary intervention: the randomized Leipzig immediate percutaneous coronary intervention abciximab IV versus IC in ST-elevation myocardial infarction trial. Circulation 118, 49–57 (2008).
- Marzilli M, Orsini E, Marraccini P, Testa R: Beneficial effects of intracoronary adenosine as an adjunct to primary angioplasty in acute myocardial infarction. Circulation 101, 2154–2159 (2000).
- Pasceri V, Pristipino C, Pelliccia F et al.: Effects of the nitric oxide donor nitroprusside on no-reflow phenomenon during coronary interventions for acute myocardial infarction. Am. J. Cardiol. 95, 1358–1361 (2005).
- Ito H, Taniyama Y, Iwakura K et al.: Intravenous nicorandil can preserve microvascular integrity and myocardial viability in patients with reperfused anterior wall myocardial infarction. J. Am. Coll. Cardiol. 33, 654–660 (1999).
- Sezer M, Oflaz H, Gören T et al.: Intracoronary streptokinase after primary percutaneous coronary intervention. N. Engl. J. Med. 356(18), 1823–1834 (2007).
- Piot C, Croisille P, Staat P et al.: Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 359(5), 473–481 (2008).
- Cura FA, L’Allier PL, Kapadia SR et al.: Predictors and prognosis of suboptimal coronary blood flow after primary coronary angioplasty in patients with acute myocardial infarction. Am. J. Cardiol. 88, 124–128 (2001).
- Brener SJ, Moliterno DJ, Aylward PE et al.: Reperfusion after primary angioplasty for ST-elevation myocardial infarction: predictors of success and relationship to clinical outcomes in the APEX-AMI Angiographic Study. Eur. Heart J. 29, 1127–1135 (2008).
- Nallamothu BK, Bradley EH, Krumholz HM: Time to treatment in primary percutaneous coronary intervention. N. Engl. J. Med. 357, 1631–1638 (2007).
- Tarantini G, Cacciavillani L, Corbetti F et al.: Duration of ischemia is a major determinant of transmurality and severe microvascular obstruction after primary angioplasty: a study performed with contrast-enhanced magnetic resonance. J. Am. Coll. Cardiol. 46(7), 1229–1235 (2005).
- Wahab NN, Cowden EA, Pearce NJ, Gardner MJ, Merry H, Cox JL: Is blood glucose an independent predictor of mortality in acute myocardial infarction in the thrombolytic era? J. Am. Coll. Cardiol. 40, 1748–1754 (2002).
- Bolk J, van der Ploeg T, Cornel JH, Arnold AE, Sepers J, Uman VA: Impaired glucose metabolism predicts mortality after a myocardial infarction. Int. J. Cardiol. 79, 207–214 (2001).
- Ceriello A: Acute hyperglycaemia: a “new” risk factor during myocardial infarction. Eur. Heart J. 26, 328–331 (2005).
- Iwakura K, Ito H, Ikushima M: Association between hyperglycemia and the no-reflow phenomenon in patients with acute myocardial infarction. J. Am. Coll. Cardiol. 41, 1–7 (2003).
- Ishihara M, Kojima S, Sakamoto T: Acute hyperglycemia is associated with adverse outcome after acute myocardial infarction in the coronary intervention era. Am. Heart J. 81, 814–820 (2005).
- Takahashi T, Hiasa Y, Ohara Y et al.: Acute hyperglycemia prevents the protective effect of pre-infarction angina on microvascular function after primary angioplasty for acute myocardial infarction. Heart 94, 1402–1406 (2008).
- Undas A, Wiek I, Stepien E: Hyperglycemia is associated with enhanced thrombin formation, platelet activation and fibrin clot resistance to lysis in patients with acute coronary syndrome. Diab. Care 31, 1590–1595 (2008).
- Iwakura K, Ito H, Kawano S et al.: Predictive factors for development of the no-reflow phenomenon in patients with reperfused anterior wall acute myocardial infarction. J. Am. Coll. Cardiol. 38, 472–477 (2001).
- Yip HK, Chen MC, Wu CL: Angiographic morphologic features of infarct-related arteries and timely reperfusion in AMI. Chest 122, 1322–1332 (2002).
- Maekawa Y, Asakura Y, Anzai T et al.: Relation of stent overexpansion to the angiographic no-reflow phenomenon in intravascular ultrasound-guided stent implantation for acute myocardial infarction. Heart Vessels 20(1), 13–18 (2005).
- Bradbury J: Renewed interest in leucocyte count as prognostic indicator after AMI. Lancet 358, 1787 (2001).
- Zalokar JB, Richard JL: Leukocyte count, smoking, and myocardial infarction. N. Eng. J. Med. 304, 465–468 (1981).
- Yarnell JW, Baker IA, Sweetnam PM et al.: Fibrinogen, viscosity and white blood cell count are major risk factors for ischemic heart disease: the Caerphilly and Speedwell collaborative heart studies. Circulation 83, 836–844 (1991).
- Barron HV, Cannon CP, Murphy SA, Braunwald E, Gibson CM: Association between white blood cell count, epicardial blood flow, myocardial perfusion and clinical outcomes in the setting of acute myocardial infarction: a thrombolysis in myocardial infarction 10 substudy. Circulation 102, 2329–2334 (2000).
- Buffon A, Biasucci LM, Liuzzo G et al.: Widespread coronary inflammation in unstable angina. N. Engl. J. Med. 347, 5–12 (2002).
- Mehta JL, Nichols WW, Mehta P: Neutrophils as potential participants in acute myocardial ischemia: relevance to reperfusion. J. Am. Coll. Cardiol. 11, 1309–1316 (1988).
- Frangogianis NG, Youker KA, Rossen RD et al.: Cytokines and microcirculation in ischemia and reperfusion. J. Mol. Cell Cardiol. 30, 2567–2576 (1998).
- Kojima S, Sakamoto T, Ishihara M et al.: The white blood cell count is an indipendent predictor of no-reflow and mortality following acute myocardial infarction in the coronary interventional era. Ann. Med. 36, 153–160 (2004).
- Sezer M, Okcular I, Umman S: Association of haematological indices with the degree of microvascular injury in patients with acute anterior wall myocardial infarction treated with primary percutaneous coronary intervention. Heart 93, 313–318 (2007).
- Zhang R, Brennan ML, FuX et al.: Association between myeloperoxidase levels and risk of coronary artery disease. JAMA 106, 2894–2900 (2001).
- Funayama H, Ishikawa S, Sugawara Y et al.: Myeloperoxidase may contribute to the no-reflow phenomenon in patients with acute myocardial infarction. Int. J. Cardiol. 139(2), 187–192 (2008).
- Van der Loo B, Martin JF: A role for changes in platelet production in the cause of acute coronary syndromes. Arterioscler. Thromb. Vasc. Biol. 19, 672–679 (1999).
- Giles H, Smith REA, Martin JF: Platelet glycoprotein IIb/IIIa and size are increased in acute myocardial infarction. Eur. J. Clin. Invest. 24, 69–72 (1994).
- Pizzuli L, Yang A, Martin JF, Luderitz B: Changes in platelet size and count in unstable angina compared with stable or non-cardiac chest pain. Eur. Heart J. 19, 80–84 (1998).
- Endler G, Klimesch A, Saunder-Plassman A et al.: Mean platelet volume is an independent risk factor for myocardial infarction but not for coronary artery disease. Br. J. Haematol. 117, 399–404 (2002).
- Pabon P, Nieto F, Morinigo JL et al.: The effect of mean platelet volume on the short-term prognosis of acute myocardial infarction. Rev. Esp. Cardiol. 51, 816–822 (1998).
- Yellon DM, Hausenloy DJ: Myocardial reperfusion injury. N. Engl. J. Med. 357, 1121–1135 (2007).
- Huczek Z, Kochman J, Filipiak KJ et al.: Mean platelet volume on admission predicts impaired reperfusion and long term mortality in acute myocardial infarction treated with primary percutaneous coronary intervention. J. Am. Coll. Cardiol. 46, 284–290 (2005).
- Niccoli G, Giubilato S, Russo E et al.: Plasma levels of thromboxane A2 on admission are associated with no-reflow after primary percutaneous coronary intervention. Eur. Heart J. 29, 1843–1850 (2008).
- Vilahur G, Casaní L, Badimon L: A thromboxane A2/prostaglandin H2 receptor antagonist (S18886) shows high antithrombotic efficacy in an experimental model of stent-induced thrombosis. Thromb. Haemost. 98(3), 662–669 (2007).
- Meier MA, Al-Badr WH, Cooper JV et al.: The new definition of myocardial infraction. Diagnostic and prognostic implications in patients with acute coronary syndromes. Arch. Intern. Med. 162, 1585–1589 (2002).
- Trevelyan J, Needham EWA, Smith SCH et al.: Sources of diagnostic inaccuracy of conventional versus new diagnostic criteria for myocardial infarction in an unselected UK population with suspected cardiac chest pain, and investigation of independent prognostic variables. Heart 89, 1406–1410 (2003).
- Jesel L, Morel O, Ohlmann P et al.: Role of pre-infarction angina and inflammatory status in the extent of microvascular obstruction detected by MRI in myocardial infarction patients treated by PCI. Int. J. Cardiol. 121, 139–147 (2007).
- Olszowska M, Tracz W: Predictive factors of myocardial reperfusion in patients with anterior wall acute myocardial infarction. Cardiol. J. 15, 57–62 (2008).
- Collinson PO, Boa FG, Gaze DC: Measurement of cardiac troponins. Ann. Clin. Biochem. 38, 423–449 (2001).
- Ohman EM, Topol EJ, Califf RM et al.: Cardiac troponin T levels for risk stratification in acute myocardial ischemia. N. Engl. J. Med. 335, 1333–1341 (1996).
- Stubbs P, Collinson P, Moseley D et al.: Prognostic significance of admission troponin T concentrations in patients with myocardial infarction. Circulation 94, 1291–1297 (1996).
- Kurowski V, Hartmann F, Killermann DP et al.: Prognostic significance of admission cardiac troponin T in patients treated successfully with direct percutaneous interventions for acute ST-segment elevation myocardial infarction. Crit. Care Med. 30(10), 2385–2387 (2002).
- Tanasijevic MJ, Cannon CP, Antman EM et al.; TIMI 10B Investigators: Myoglobin, creatine-kinase-MB and cardiac troponin-I 60-minratios predict infarct-related artery patency after thrombolysis for acute myocardial infarction: results from the thrombolysis in myocardial infarction study (TIMI) 10B. J. Am. Coll. Cardiol. 34, 739–747 (1999).
- Stewart JT, French JK, Theroux P et al.: Early noninvasive identification of failed reperfusion after intravenous thrombolytic therapy in acute myocardial infarction. J. Am. Coll. Cardiol. 31, 1499–1505 (1998).
- Matetzky S, Sharir T, Domingo M et al.: Elevated troponin I level on admission is associated with adverse out come of primary agioplasty in acute myocardial infarction. Circulation 102, 1611–1616 (2000).
- Giannitsis E, Müller-Bardorff M, Lehrke S et al.: Admission troponin T level predicts clinical outcomes, TIMI flow and myocardial tissue perfusion after primary percutaneous intervention for acute ST-segment elevation myocardial infarction. Circulation 104, 630–635 (2001).
- Hong SN, Ahn Y, Hwang SH et al.: Usefulness of preprocedural N-terminal pro-brain peptide in predicting angiographic no-reflow phenomenon during stent implantation in patients with ST-segment elevation acute myocardial infarction. Am. J. Cardiol. 100, 631–634 (2007).
- Porto I, Burzotta F, Trani C et al.: Elevated admission cardiac troponin T is associated with microvascular dysfunction in acute myocardial infarction treated with emergency angioplasty. J. Cardiovasc. Med. (Hagerstown). 10(8), 664–668 (2009).
- Libby P, Ridker PM, Maseri A: Inflammation and atherosclerosis. Circulation 105, 1135–1143 (2002).
- Tomai F, Ribichini F, Ghini AS et al.: Elevated C-reactive protein levels and coronary microvascular dysfunction in patients with coronary artery disease. Eur. Heart J. 26, 2099–2105 (2005).
- Sturk A, Hack CE, Aarden LA, Brouwer M, Koster RR, Sanders GT: Interleukin-6 release and the acute-phase reaction in patients with acute myocardial infarction: a pilot study. J. Lab. Clin. Med. 119, 574–579 (1992).
- Speidl WS, Zeiner A, Nikfardjam M et al.: An increase of C-reactive protein is associated with enhanced activation of endogenous fibrinolysis at baseline but an impaired endothelial fibrinolytic response after venous occlusion. J. Am. Coll. Cardiol. 45, 30–34 (2005).
- Lagrand WK, Niessen HW, Wolbink GJ et al.: C-reactive protein colocalizes with complement in human hearts during acute myocardial infarction. Circulation 95, 97–103 (1997).
- De Sutter J, De Buyezere M, Gheeraert P et al.: Fibrinogen and C-reactive protein on admission as marker of final infarct size after primary angioplasty for acute myocardial infarction. Atherosclerosis 157, 189–196 (2001).
- Théroux P, Armstrong PW, Mahaffey KW et al.: Prognostic significance of blood markers of inflammation in patients with STsegment elevation myocardial infarction undergoing primary angioplasty and effects of pexeluzimab, a C5 inhibitor: a substudy of the COMMA trial. Eur. Heart J. 26(19), 1964–1970 (2005).
- Qamirani E, Ren Y, Kuo L, Hein TW: C-reactive protein inhibits endotheliumdependent NO-mediated dilation in coronary arterioles by activating p38 kinase and NAD(P)H oxidase. Arterioscler. Thromb. Vasc. Biol. 25, 995–1001 (2005).
- Celik T, Iyisoy A, Kursaklioglu H et al.: The impact of admission C-reactive protein levels on the development of poor myocardial perfusion after primary percutaneous intervention in patients with acute myocardial infarction. Coron. Artery Dis. 16, 293–299 (2005).
- Niccoli G, Lanza GA, Spaziani C, Crea F: Baseline sistemi inflammatory status and no-reflow phenomenon after percutaneous coronary angioplasty for acute myocardial infarction. Int. J. Cardiol. 117, 306–311 (2007).
- Wasilewski J, Turczynski B, Kowalik V et al.: Evaluation of hemorpheological profiles in patients with no-reperfusion phenomenon in acute ST elevation myocardial infarction treated by primary percutaneous coronary intervention. J. Vasc. Biol. 42(Suppl. 2), 49 (2005).
- Soutani M, Suzuki Y, Tateishi N et al.: Quantitative evaluation of flow dynamics of erythrocytes in microvessels: influence of erythrocyte aggregation. Am. J. Physiol. 268, H1959–H1965 (1995).
- De Sutter J, De Buyzere M, Gheeraert P et al.: Fibrinogen and C-reactive protein on admission as markers of final infarct size after primary angioplasty for acute myocardial infarction. Atherosclerosis 157, 189–196 (2001).
- Wasilewski J, Osadnik T, Polonski L: High baseline fibrinogen concentration as a risk factor of no tissue reperfusion in ST-segment elevation acute myocardial infarction treated with successful primary percutaneous coronary intervention. Kardiol. Pol. 64, 967–972 (2006).
- Sgueglia GA, Niccoli G, Spaziani C, Crea F et al.: Baseline von Willebrand factor plasma levels and no-reflow phenomenon after primary percutaneous coronary intervention for ST segment elevation myocardial infarction. Int. J. Cardiol. (2009) (Epub ahead of print).
- Yanagisawa M, Kurihara H, Kimura S et al.: A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 332, 411–415 (1988).
- Resink TJ, Hahn AW: Scott-Burden T, Powell J, Weber E, Buhler FR: Inducible endothelin mRNA expression and peptide secretion in cultured human vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 168, 1303–1310 (1990).
- Winkles JA, Alberts GF, Brogi E, Libby P: Endothelin 1 and endothelin receptor mRNA expression in normal and atherosclerotic human arteries. Biochem. Biophys. Res. Commun. 191, 1081–1088 (1993).
- Molenaar P, O’Reilly G, Sharkey A et al.: Characterization and localization of endothelin receptor subtypes in the human atrioventricular conducting system and myocardium. Circ. Res. 72, 526–538 (1993).
- Sugden PH, Bogoyevitch MA: Endothelin-ldependent signaling pathways in the myocardium. Trends Cardiovasc. Med. 6, 87–94 (1996).
- Hasdai D, Kornowski R, Battler A: Endothelin and myocardial ischemia. Cardiovasc. Drugs Ther. 8, 589–599 (1994).
- Grover GJ, Dzwonczyk S, Parham CS: The endothelin-1 receptor antagonist BQ-123 reduces infarct size in a canine model of coronary occlusion and reperfusion. Cardiovasc. Res. 27, 1613–1618 (1993).
- Krause SM, Lynch JJ, Stabilito II et al.: Intravenous administration of the endothelin-1 antagonist BQ-123 does not ameliorate myocardial ischaemic injury following acute coronary artery occlusion in the dog. Cardiovasc. Res. 28, 1672–1678 (1994).
- Galiuto L, DeMaria AN, Iliceto S: Ischemiareperfusion injury at the microvascular level: treatment by endothelin A-selective antagonist and evaluation by myocardial contrast echocardiography. Circulation 102, 3111–3116 (2000).
- Basso C, Thiene G, Della Barbera M et al.: Endothelin A-receptor antagonist administration immediately after experimental myocardial infarction with reperfusion does not affect scar healing in dogs. Cardiovasc. Res. 55, 113–121 (2002).
- Niccoli G, Lanza GA, Shaw S, Crea F et al.: Endothelin-1 and acute myocardial infarction: a no-reflow mediator after successful percutaneous myocardial revascularization. Eur. Heart J. 27, 1793–1798 (2006).
- Adlbrecht C, Bonderman D, Plass C et al.: Active endothelin is an important vasoconstrictor in acute coronary thrombi. Thromb. Haemost. 97(4), 642–649 (2007).
- Edgington TS, Mackman N, Brand K, Ruf W: The structural biology of expression and function of tissue factor. Thromb. Haemost. 66, 67–79 (1991).
- Carson SD, Brozna JP: The role of tissue factor in the production of thrombin. Blood Coagul. Fibrinolysis 4, 281–292 (1993).
- Mallat Z, Hugel B, Ohan J, Leseche G, Freyssinet JM, Tedgui A: Shed membrane microparticles with procoagulant potential in human atherosclerotic plaques: a role for apoptosis in plaque thrombogenicity. Circulation 99, 348–353 (1999).
- Bonderman D, Teml A, Jakowitsch J et al.: Coronary no-reflow is caused by shedding of active tissue factor from dissected atherosclerotic plaque. Blood. 99(8), 2794–2800 (2002).
- James SK, Lindahl B, Siegbahn A et al.: N-terminal pro-brain natriuretic peptide and other risk markers for the separate prediction of mortality and subsequent myocardial infarction in patients with unstable coronary artery disease. Circulation 108, 275–281 (2003).
- Omland T, de Lemos JA, Morrow DA et al.: Prognostic value of N-terminal pro-atrial and probrain natriuretic peptide in patients with acute coronary syndromes. Am. J. Cardiol. 89, 463–465 (2002).
- Goetze JP, Christoffersen C, Perko M et al.: Increased cardiac BNP expression associated with myocardial ischemia. FASEB J. 17, 1105–1107 (2003).
- Grabowski M, Filipiak KJ, Karpinski G: Serum B-type natriuretic peptide levels on admission predict not only short-term death but also angiographic success of procedure in patients with acute ST-elevation myocardial infarction treated with primary angioplasty. Am. Heart J. 148, 655–662 (2004).
- Jeong YH, Kim WJ, Park DW: Serum B-type natriuretic peptide on admission can predict the ‘no-reflow’ phenomenon after primary drug-eluting stent implantation for ST-segment elevation myocardial infarction. Int. J. Cardiol. (2009) (Epub ahead of print).