Review Article - Imaging in Medicine (2012) Volume 4, Issue 2
An update on brain imaging in parkinsonian dementia
Myria Petrou*, Vikas Kotagal & Nicolaas I BohnenUniversity of Michigan, Ann Arbor, MI, USA
- Corresponding Author:
- Myria Petrou
University of Michigan, Ann Arbor, MI, USA
Tel: +1 734 615 3586
Fax: +1 734 764 2412
E-mail: mpetrou@med.umich.edu
Abstract
Disturbances of cognition are frequent in Parkinson’s disease (PD). Unlike severe loss of dopamine early in PD, extensive cholinergic losses have been consistently reported in PD with dementia. Cholinergic imaging suggests that basal forebrain cholinergic system degeneration appears early in PD and worsens with dementia development. Cortical cholinergic denervation is similar in PD with dementia and dementia with Lewy bodies, supporting a common disease spectrum, at least with respect to cholinergic pathology. Presence of cerebral amyloidopathy in the setting of parkinsonism may accelerate cognitive decline. Novel MRI techniques illustrate the widespread presence of neurodegeneration in PD with dementia, affecting white matter tracts and connectivity functions. This review will outline current concepts regarding dementia development in PD and discuss their correlation with functional and structural neuroimaging including PET and MRI.
Keywords
acetylcholine ▪ amyloid ▪ dementia with Lewy bodies ▪ fluorodeoxyglucose ▪ MRI ▪ Parkinson’s disease ▪ PET ▪ SPECT
Cognitive dysfunction in Parkinson’s disease
Disturbances of cognition are frequent findings in Parkinson’s disease (PD) patients with point prevalence estimates of frank dementia (PD with dementia [PDD]) being reported in 40% and more subtle cognitive impairment affecting over 60% of PD patients [1–3]. Older age and duration of disease are associated with increased risk of dementia, with development of dementia in 75% and over 80% of PD patients who survive for more than 10 and 20 years, respectively [4,5].
An arbitrary but generally accepted distinction is made using current consensus diagnostic criteria between patients presenting with parkinsonism prior to the onset of dementia (PDD) versus those with concurrent development of parkinsonism and dementia or with dementia preceding parkinsonism (dementia with Lewy bodies [DLB]). Using the so-called ‘1-year rule’, patients with L-Dopa responsive parkinsonism who develop dementia more than 1 year after their initial PD motor symptoms are classified as PDD. Patients with onset of dementia and parkinsonism within 1 year or with dementia preceding parkinsonism are classified as DLB [6].
Mild selective cognitive deficits in PD are frequently present in the absence of a clinical diagnosis of dementia [3]. Although impairments of executive functions have long been considered the cognitive signature of mild cognitive impairment (MCI) in PD, it is now widely recognized that cognitive impairments in early PD are heterogeneous, with a number of patients exhibiting deficits on posterior cortically based cognitive tasks. For example, the meta-analysis by Aarsland et al. on MCI in PD suggested that memory and visuospatial impairments were more common than executive impairment in nondemented PD [7]. MCI in PD may represent a precursor state to dementia but is qualitatively distinct from the amnestic-type MCI seen in association with Alzheimer’s disease (AD). In this respect, the longitudinal study by Williams-Gray et al. suggests that posterior cortical deficits herald the dementia of PD [8].
The mixed pattern of cognitive decline in PD is probably secondary to a combination of different neuropathological processes, which worsen and become more complex and intertwined with disease progression. The pathological hallmark of parkinsonian dementia is the presence of extranigral Lewy bodies [9,10], but can be accompanied by other pathologies, such as AD-type findings [11], including b-amyloid plaques and hyperphosphorylated tau tangle pathology. In addition, comorbid vascular pathology, manifesting as leukoaraiosis on imaging studies, may also contribute to PDD [12,13]. These different neuropathological changes may not only result in local or regional neuronal and/or synaptic dysfunction but can also interrupt neuromodulator projection networks, including the cholinergic, noradrenergic and dopaminergic systems.
This review will: discuss current evidence and theory on the etiology of cognitive impairment and dementia in PD; outline alterations in cerebral blood f low (CBF) and glucose metabolism in PDD using SPECT, and [18F] Fluorodeoxyglucose (FDG) PET and MR perfusion techniques; review brain PET imaging findings regarding abnormalities in the dopaminergic and cholinergic systems, as well as amyloid deposition in the setting of PD-related cognitive impairment; and provide an overview of MR findings in PD-associated cognitive impairment.
Post-mortem findings of parkinsonian dementia
There are numerous pathologic changes in PDD dementia, including degeneration of subcortical dopaminergic and cholinergic projection systems, as well as direct cortical involvement. Cortical Lewy bodies and Lewy neurites as well as AD-type changes with b-amyloid plaques, and in some instances neurofibrillary tangles, are present in PDD subjects [14–20]. Some neuropathology studies suggest that limbic and neocortical Lewy body deposition is the main determinant of cognitive decline in PD [10,21–24], whereas others suggest an important role for amyloid plaques and neurofibrillary pathology [25–27]. Striatal amyloid deposition is also reported in the setting of PD and cognitive impairment [20,28]. Interestingly, a recent post-mortem study found a greater frequency of striatal b-amyloid deposition in PDD compared with PD [28], and may thus confer increased risk of dementia. A recent pathologic study by Compta et al. suggests that the combination of Lewy bodies, amyloid plaques and neurofibrillary tangles is the crucial determinant of dementia in PD [29]. The complex interplay between amyloid deposition and Lewy body pathology may become clearer in the era of in vivo fibrillary amyloid imaging. In vitro evidence suggests an interaction between PD-type (a-synuclein) and AD-type (i.e., tau) pathologies [30].
Loss of mesencephalic dopaminergic neurons in the substantia nigra pars compacta and nerve terminals in the striatum is the pathological hallmark of PD. Significant nigrostriatal dopaminergic cell loss occurs early in symptomatic PD. Its severity and universal prevalence early in the disease course may explain why subsequent emergence and worsening of non-motor symptoms are increasingly attributed to nondopaminergic degenerations in advancing PD. Despite this, post-mortem cell loss in the medial substantia nigra is seen more commonly in PDD as opposed to PD without dementia, suggesting an association between dementia, duration of disease and the integrity of nigrostriatal dopaminergic pathways [31]. Striatal a-synuclein pathology is also seen more commonly in PDD than in PD without dementia or DLB [32,33]. Although duration of disease may be a confounding factor in PD to account for the more severe striatal pathology, this argument may be less valid in DLB.
In addition to nigrostriatal denervation, there is evidence for alterations in cholinergic neurotransmission in PD. Loss of cholinergic neurons in the nucleus basalis of Meynert has been reported in both PD and AD brains [34–39]. Interestingly, neuronal forebrain loss may be even more profound in PD compared with AD [34].Similarly, a post-mortem study found greater reductions of acetylcholinesterase (AChE) in the frontal cortex of PDD compared with nondemented patients with PD [40]. The reduction in cortical nicotinic cholinergic receptor number in PD parallels the degree of dementia observed with progression of the disease [41,42]; it is hence believed that degeneration of the cholinergic system may play a significant role in the development of dementia in PD.
Imaging global cerebral cortical dysfunction in parkinsonian dementia: FDG-PET & CBF imaging
FDG-PET and [99m]Tc-ECD, [99m]Tc-HMPAO or [123I]-iodoamphetamine-SPECT can be used to estimate cerebral glucose metabolism and CBF, both of which serve as surrogate markers for regional cortical health and metabolism in neurodegenerative diseases. More recently, Arterial spin labeling (ASL) MRI has been used to study perfusion changes [43]. Each of these techniques has relative advantages and disadvantages in the assessment of cerebral perfusion and/or metabolism. FDG-PET is both precise and accurate but can be expensive and time consuming to perform. SPECT is less costly but offers poorer spatial resolution. ASL does not involve the use of ionizing radiation or contrast injection but does carry potential for a low signal-to-noise ratio.
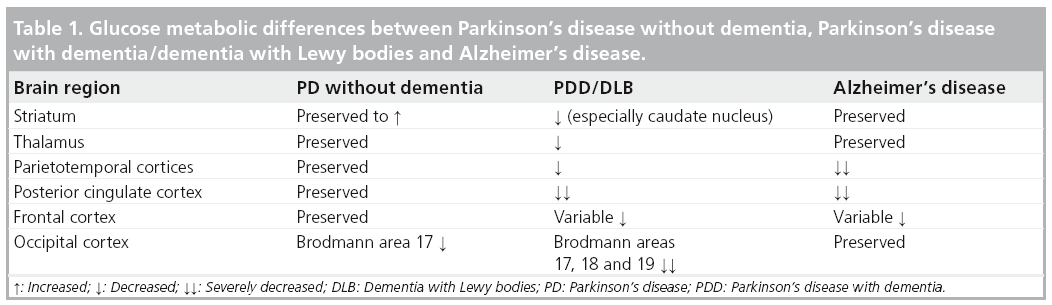
FDG-PET has been studied extensively in AD where it shows characteristic parietotemporal and posterior cingulate glucose hypometabolism [44,45]. While similar findings of posterior cingulate hypometabolism are also seen in PDD, the presence of anterior cingulate hypometabolism may be more characteristic of DLB than PDD [45,46]. Our group recently reported results of a prospective study involving FDG-PET in 23 nondemented individuals with PD and 27 controls [47]. The six out of 23 PD subjects who went on to develop incident clinical dementia by the 2–6-year clinical followup displayed significant baseline reductions in FDG-PET metabolism in the occipital–visual association cortex (especially, Brodmann area 18) and posterior cingulate cortex (Figure 1). Early involvement of the visual association cortex is interesting given the prominent visual hallucinations seen in advanced PDD. Mild metabolic reductions were also seen in the caudate nucleus.

Figure 1: Metabolic reduction in Parkinson’s disease. 3D-SSP t-statistic maps
comparing PD dementia nonconverters and converters based on voxel-based
comparison to normative data from healthy controls at baseline. The most
prominent metabolic reduction in the prospective PDD converters is evident in the
cuneus (especially Brodmann area 18) and precuneus. Mild-to-moderate reductions
are also present in the mesiofrontal lobes. In addition, the PD dementia converter
subjects demonstrate relative sparing of the primary sensorimotor cortex, a pattern
similar to Alzheimer’s disease. In PD nonconverter subjects, metabolic reduction in
the posterior calcarine cortex (Brodmann area 17) is evident, while there is relative
sparing of Brodmann areas 18, 19 and the precuneus.
LLAT: Left lateral; LMED: Left medial; PD: Parkinson’s disease; PDD: Parkinson’s
disease with dementia; RLAT: Right lateral; RMED: Right medial.
In five out of six subjects who developed dementia, repeat FDG-PET imaging after 2 years showed significant interval changes in the thalamus (-11.4%), posterior cingulate (-9%), occipital (-7%), parietal (-7%) and frontal cortices (-7%), and mild reductions in temporal cortices (-5%) and hippocampi (-3%) compared with study entry scans [47]. An example of serial FDG-PET imaging over 4 years in a PD subject converting from MCI to dementia is shown in Figure 2.

Figure 2: Progressive metabolic reduction from minimal cognitive
impairment to clinical diagnosis of dementia in a single patient. Voxel-based
statistical maps demonstrate regional cerebral glucose metabolic reductions relative
to a database of normal subjects. Progressive metabolic reductions in the cuneus
and precuneus occur before less severe, but more widespread, cortical reductions.
Sparing of the primary sensorimotor cortical strip is present.
LLAT: Left lateral; LMED: Left medial; RLAT: Right lateral; RMED: Right medial.
These findings are in agreement with a cross-sectional study by Jokinen et al. who also reported occipital FDG hypometabolism in PD without dementia compared with more extensive cortical, thalamic and caudate nucleus hypometabolism in PDD subjects [48]. Findings of mixed cortical and subcortical pathology on imaging are also consistent with the current paradigm of mixed pathological substrates leading to dementia in the context of PD (Table 1) [49]. Additionally, the presence of both posterior (parieto–occipital) and anterior cortical (frontal) changes may reflect distinct cognitive syndromes in PD and cognitive impairment. For example, Williams-Gray and colleagues reported that more posterior cortically based cognitive defects evolved into later dementia, whereas frontostriatal executive deficits were not associated with subsequent dementia risk [8].
Regional CBF (rCBF) can be estimated using SPECT imaging and may help differentiate between different types of dementia. Sawada et al. found no significant difference between rCBF in nondemented subjects with PD and normal controls in a [123I]-iodoamphetamine-SPECT study [50], but did report bifrontal and biparietal hypometabolism in PDD subjects compared with healthy controls. Biparietal CBF was most strongly associated with poor selective performance on neuropsychological testing. These findings are in agreement with other studies and suggest that the pattern of rCBF seen in PDD is relatively similar to AD [51,52]. rCBF studies may also differentiate DLB from AD on the basis of occipital hypoperfusion in DLB [53]. Increased severity of frontal rCBF abnormalities can also be seen in DLB compared with AD [54], although DLB with motor symptoms of parkinsonism is associated with reduced rCBF in primary motor and supplementary motor cortices compared with DLB without parkinsonism [55]. These findings all suggest that DLB may manifest with a regionally distinct pattern of cortical blood flow alterations compared with AD.
Kamagata et al. recently used ASL-MRI to study posterior perfusion changes in a small group of PDD subjects compared with PD subjects and healthy controls. They found significant reductions in rCBF in the posterior cortex (occipital, inferolateral and medial parietal) in PDD subjects compared with PD subjects who in turn had significantly lower posterior cortical rCBF compared with healthy controls [43]. As this study did not examine other cerebral regions of interest, it remains unclear whether their disease-specific ASL-MRI findings may have been equally severe or even more profound in other cortical regions of interest.
Imaging neurochemical changes in parkinsonian dementia: dopamine & cholinergic imaging
Degeneration in dopamine projection systems within the CNS can be assessed in vivo using dopamine SPECT and PET imaging techniques. PET methods include the use of [18F]-fluorodopa (FDOPA)-PET along with other measures of dopaminergic terminal integrity, such as the presynaptic dopamine transporter (DAT; e.g., [11C]-methylphenidate) and the monoaminergic ligand [11C]-dihydrotetrabenazine. Dihydrotetrabenazine is a vesicular monoamine transporter type 2 (VMAT2) that binds to VMAT2 terminals in the striatum and serves as a reliable marker for dopamine terminal integrity. SPECT methods include DAT tracers, such as [123I]-2b-carbomethoxy-3b-(4- iodophenyl)-N-(3-fluoropropyl)-N-nortropane ([123I]-FP-CIT) and [23I]-2b-carbomethoxy- 3b-(4-iodophenyl)-N-(3-fluoropropyl) nortropane ([I123]-b-CIT). [123I]-FP-CIT (known as Ioflupane or DaTscan™) has been approved to assist in the clinical evaluation of patients with parkinsonism and its distinction from essential tremor. The ligand is also approved in Europe to assist with the clinical diagnosis of DLB in its distinction from prototypical AD.
Similar to imaging findings in PD without dementia, PDD and DLB show reduced presynaptic dopaminergic tracer uptake in the striatum [56,57]. Figure 3 shows VMAT2 binding losses in patients with PD, PDD and DLB as determined by [11C]-dihydrotetrabenazine PET.

Figure 3: Vesicular monoamine transporter type 2 binding images. (A) Binding in a normal subject. (B) Binding in a patient with early Parkinson’s disease without dementia who has predominant asymmetric loss. (C) Parkinson’s disease with dementia and (D) dementia with Lewy bodies as determined by [11C]-dihydrotetrabenazine PET. More significant striatal losses are present in the Parkinson’s disease with dementia and dementia with Lewy bodies subjects.
The caudate nucleus and associated ventral striatum are thought to play a more significant role in cognition in comparison to the more motor-related putamen [58]. These differential roles are supported by dopaminergic studies in PD showing that poor performance on memory and executive functioning is closely correlated to reduced FDOPA tracer uptake in the caudate [59]. Interestingly, poor performance on attentional measures, such as the Stroop test, have been shown to correlate negatively with an increased FDOPA-PET signal in the medial frontal cortex and anterior cingulate in drugnaive PD subjects. A possible explanation is that extrastriatal, frontal lobe dopaminergic systems may also be modulated in early PD [60], probably based on a compensatory mechanism [61].
Cross-sectional dopaminergic studies have failed to show a difference between PDD and DLB in nigrostriatal dopaminergic denervation [62]. Similarly, a longitudinal DAT-SPECT imaging study revealed similar rates of decline between PDD and DLB over time [63]. This study also found that dementia and motor symptom severity correlated with dopaminergic decline, suggesting that dopaminergic loss may play an important role in cognitive as well as motor function in the context of PD. While nigrostriatal dopaminergic denervation is a near-universal phenomenon in PD with and without dementia, it occurs early in PD and thus cannot completely account for the subsequent full-blown development of dementia.
Post-mortem studies have shown significant cholinergic losses in PDD compared with PD without dementia [34,40]. Vesicle-associated acetylcholine transporters (VAChT) are responsible for transporting acetylcholine into intracellular vesicles from the cytoplasm and, when radiolabelled in vivo, can provide a surrogate marker of presynaptic terminal density. There are several radioligands that target the VAChT [64], but only one of these, [(−)-5-123I]-iodobenzovesamicol ([123I]-IBVM), a SPECT radiotracer, has been used to image the living human brain [64]. [123I]-IBVM, an analog of vesamicol, binds to the acetylcholine vesicular transporter and has a demonstrated relative distribution in the human brain, which corresponds well with postmortem values reported for choline acetyltransferase [64]. A potential clinical advantage of the VAChT ligand is that it may allow in vivo assessment of integrity of cholinergic nerve terminals despite patients taking cholinesterase inhibitor drugs. Radioligands have also been developed to measure cholinergic enzymes, including AChE localized in both pre- and post-synaptic targets. Cholinergic receptor ligands are selective for either muscarinic or nicotinic receptors.
Previous in vivo imaging studies have shown reduced cortical AChE and VAChT activity in PDD and DLB compared with PD without dementia [56,65–67].
Previous VAChT imaging studies have reported cholinergic def icits in PD and PDD patients [65]. In PD without dementia, [123I]-IBVM binding was reduced modestly in the average cortex (-9%) with more extensive reductions noted in the parietal (-19%) and occipital (-21%) cortices. Demented PD subjects had extensive cortical binding decreases that were most prominent in the occipital (-39%) and posterior cingulate (-45%) cortices. Furthermore, cortical VAChT losses were greater in PDD compared with AD of the lateonset type and comparable to AD of early onset [65]. Using AChE-PET, we also reported that in vivo cortical AChE levels were more severe and widespread in PDD compared with AD of similar dementia severity [66].
Figure 4 shows AChE-PET imaging findings in subjects with PDD and DLB.

Several studies have correlated performance on cognitive tests with cortical cholinergic activity. Shinotoh et al. noted significantly lower cortical AChE activity in PD subjects with visual hallucinations but did not find a correlation between cortical AChE activity and scores on the Mini-Mental State Examination (MMSE) or the Wisconsin Card Sorting Test in a predominantly nondemented PD population [68]. We recently reported a modest inverse correlation between cortical AChE activity and MMSE scores in a larger series of PD subjects without dementia (r = 0.36, p = 0.02) [69]. In a different study, we found that performance on the WAIS-III Digit Span, a test of working memory and attention, correlated well with cortical AChE activity (r = 0.61, p < 0.005) in a combined study of PD, PDD and DLB patients. There were also significant correlations between cortical AChE activity and additional tests of attentional and executive functions, such as the Trail Making Test and Stroop Color Word Test [70]. No significant correlations were seen between AChE activity and delayed verbal learning in this study. Shimada et al. found an inverse correlation between cortical AChE activity and MMSE scores in a combined PD/PDD/ DLB cohort (r = -0.47, p < 0.005) [67]. Subgroup analysis revealed a more robust inverse correlation between posterior cingulate AChE activity and MMSE scores in the DLB subjects in this study [67].
These findings suggest that attentional and executive cognitive domains in PD may be regulated in part by cholinergic projection neurons arising from the basal forebrain. This theory is corroborated by observational data on anticholinergic medications, which have been shown to specifically worsen performance on tests of executive function and attention in PD, including the Digit Span test and the Wisconsin Card Sorting Task [71–74]. PD patients may also have a disease-specific vulnerability to the harmful cognitive side effects of anticholinergic medications [74,75]. Taken together, these data suggest that the ‘cholinergic model’ classically associated with AD may provide an even more robust explanation of progressive cognitive dysfunction in the setting of PD. Cognitive impairment in PD is probably the result of a combination of pathologic changes that include degeneration of the dopaminergic, noradrenergic and cholinergic systems. For example, Klein et al. did not observe a difference in striatal FDOPA reductions between PD and PDD groups, whereas the PDD group did show a greater reduction in cortical AChE compared with the PD group without dementia (Table 2) [76]. These findings confirm the current view that, unlike early stage PD, where there is evidence of uniform and severe dopaminergic denervation, the appearance of dementia in PD is associated with progressive cholinergic denervation. These findings are also concordant with reports of MRI volumetric measurements of the substantia innominata showing mild volume loss in early stage PD but more profound losses in PDD [77].

Imaging proteinopathies: amyloidopathy
The recent developments of radioligands that target fibrillary b-amyloid offer a novel opportunity to study in vivo amyloid protein aggregation [78,79]. 11C-Pittsburgh compound B (PiB) is a widely used PET b-amyloid ligand; studies with 11C-PiB show robust neocortical binding in AD subjects [78,80].
Previous in vivo imaging studies indicate lower levels of neocortical PiB binding in PD and PDD patients when compared with AD patients [81–85]. By contrast, patients with DLB may exhibit elevated, but variable, neocortical PiB binding, an interesting observation as the distinction between PDD and DLB is made on clinical grounds based on the relative timing of the onset of dementia and parkinsonism [84,85]. For example, Edison et al. found evidence of elevated cortical PiB binding in most DLB subjects but activity levels were generally lower and more variable compared with AD [84]. They found high global cortical amyloid burden in DLB but low global cortical amyloid burden in PDD subjects. By contrast, Maetzler and colleagues found abnormal cortical PiB binding in two out of ten PDD subjects [86]. Foster et al. used b-amyloid PET imaging to examine subjects with a variety of Lewy body disorders, including DLB (n = 6), PD (n = 8) and PDD (n = 15), and found no significant differences between any of these disease groups in average PiB binding in any cortical regions of interest [81]. A post-mortem case series of three patients with PDD who had in vivo PiB imaging found elevated cortical PiB ligand uptake in two of the three patients. At autopsy, all three individuals had abundant cortical Lewy bodies and were classified as low-probability AD based on National Institute on Aging-Reagan criteria [87]. The two PiB-positive individuals had abundant diffuse b-amyloid plaques but only sparse neuritic plaques and intermediate neurofibrillary tangle pathology. The PiB-negative individual had rare diffuse plaques, no neuritic plaques and low neurofibrillary tangle burden. These data indicate that PiB-PET is specific for fibrillar b-amyloid molecular pathology but not for pathologic diagnosis of comorbid AD in patients with PDD. Findings of generally higher global cortical amyloid burden in DLB but lower global cortical amyloid burden in PDD suggest that cortical fibrillary b-amyloid deposition is not a requisite for PD dementia but may differentially contribute to the temporal manifestation and/or nature of the neurobehavioral phenotype of parkinsonian dementia. Whether a combination of amyloid and Lewy body pathology exacerbates the memory and cognitive problems in DLB is unclear but it is likely that it accelerates the dementia process, whereas Lewy body pathology alone may lead to a slower dementing process in PDD [25].
Figure 5 shows PiB-PET findings in a patient with PD (no abnormal binding) and abnormal elevated binding in a PDD subject.

Figure 5: 11C-Pittsburgh compound B PET Images. (A) Findings in a patient with Parkinson’s disease with no abnormal binding. (B) abnormal elevated binding in a Parkinson’s disease with dementia subject.
Structural MRI findings in parkinsonian dementia
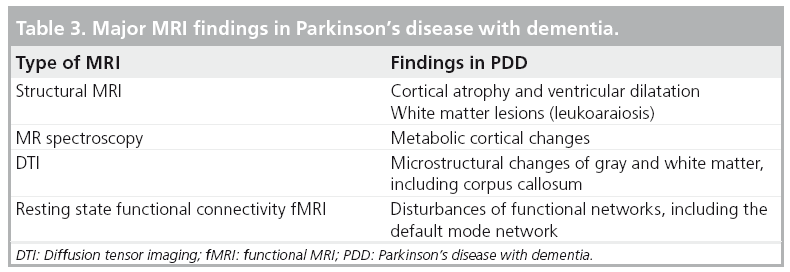
A number of MRI studies have reported cortical atrophy in PD. Studies using a region of interest approach have demonstrated atrophy of the hippocampus and the amygdala in PD patients with and without dementia [88–91]. Furthermore, studies have shown correlations between atrophy in these structures and cognitive performance in PD patients [89–93]. Voxel-based morphometry (VBM), a fully automated whole-brain measurement technique that maps out the statistical probability of differences in regional tissue volume or density between groups has also been used in the study of PD patients. VBM brain MRI studies have shown that PDD subjects present with limbic and widespread neocortical gray matter loss; PD subjects without dementia mainly present with atrophy in the frontal and temporal regions [94–97]. Nondemented PD subjects at higher risk for dementia also show greater atrophy than PD subjects without these factors. For example, Beyer et al. found that patients with PD and MCI had gray matter reductions in temporal and frontal areas when compared with patients without MCI [97]. Nondemented patients with visual hallucinations (and therefore at increased risk for dementia) were also found to have greater occipitoparietal gray matter loss compared with PD patients without hallucinations [96]. A longitudinal study employing ventricular volume as a measure of global brain volume loss showed greater ventricular volumes in PD subjects who developed dementia within a 36-month period versus PD subjects who remained cognitively intact [98]. Furthermore, there was a significant correlation between ventricular volume change and dementia rating scale scores over the 36-month period. Hu et al. also found that annual brain volume loss is greater in PD patients compared with controls and that these changes correlated with cognitive decline [99]. Burton et al. found higher rates of atrophy in PDD subjects on serial MRI compared with nondemented PD subjects and controls; there was, however, no difference in the rate of atrophy between nondemented PD subjects and healthy controls [100]. A VBM-based study in PDD reported limbic and temporo–occipital areas of gray matter reduction after a 25-month follow-up compared to baseline imaging [94]. Figure 6 shows an example of global atrophy and dilated ventricles in a PDD subject.

Figure 6: Transaxial MRI fluid-attenuated inversion recovery image showing global atrophy and enlarged ventricles in a Parkinson’s disease with dementia subject.
White matter disease is an area of increasing interest in PD. Leukoaraiosis or white matter lesions (WML) are commonly observed on imaging studies in the elderly and may present as signal hyperintensities on T2-weighted MRI studies. As age-associated WML are associated with cognitive deficits in otherwise normal elderly individuals [101–104], it is plausible that WML may also contribute to cognitive symptoms in PD [12,13,70,105]. For example, Lee and colleagues found that PDD patients had significantly more supratentorial WML, particularly periventricular WML, than the PD group [106]. By contrast, other studies have failed to find significant disease-specific cognitive correlates of WML, at least in early stage disease [107,108]. It is also possible that cognitive effects of comorbid WML may be more prominent in advancing disease where presence of multiple neurodegenerations are expected to lower the clinical symptomatic threshold for a single pathological process owing to loss of cerebral reserve capacity.
MR spectroscopy
MR spectroscopy is an advanced MR technique that can provide a noninvasive insight into brain biochemistry and has been used to study PDD. Bowen et al. reported elevated lactate levels in the occipital region in PDD subjects compared with nondemented PD patients and healthy controls; this study found no differences in N-acetylaspartate (NAA), which is considered a neuronal marker [109]. Summerfield et al. reported lower NAA levels in the occipital cortex of PDD subjects compared with PD subjects without dementia but no difference between PDD subjects and healthy controls [110]. A more recent study looking at the posterior cingulate gyrus with MR spectroscopy reported lower NAA:creatine (Cr) ratios in PDD subjects compared with healthy controls and to PD subjects without dementia [111]. MR spectroscopy abnormalities in PDD and AD were different. Both PDD and AD subjects showed a reduced NAA:Cr in the posterior cingulate. PDD subjects also showed reduced glutamate:Cr in the same region compared with both controls and AD subjects. AD subjects showed increased choline (Cho):Cr and elevated myoinositol compared with healthy controls, a finding that was not seen in PDD subjects [112].
Diffusion tensor imaging
Diffusion tensor imaging (DTI) is an MRI technique that examines the local microstructural characteristics of water diffusion and is used to evaluate the integrity of white matter fiber tracts. DTI measures of the magnitude and direction of water diffusion are termed mean diffusivity (MD) and fractional anisotropy (FA) where MD is a measure of water diffusivity and FA of tract directionality and integrity [113]. There is little data regarding DTI changes in PDD. A DTI study by Matsui et al. employed region of interest analysis and showed reduced FA values in frontal, temporal and occipital white matter in both PD and PDD patients [114]. Reduced FA values were also noted in the posterior cingulate white matter of PDD versus PD patients. A study of DLB subjects showed decreased FA and increased MD in the corpus callosum and pericallosal regions [115]. A similar study in PDD and nondemented PD subjects did not show any group differences in MD and FA values of the corpus callosum and cingulum. There was, however, significant correlation between MD values and MMSE scores [116].
Functional MRI techniques
Functional MRI (fMRI) is based on measuring and analyzing the blood-oxygen-level-dependent (BOLD) effect. While the exact relationship between neural activity and changes in BOLD signal are still under investigation, the BOLD signal appears to be indirectly linked to neural activity [117]. There are no task-based fMRI studies in PDD subjects, presumably owing to difficulties with task execution in the MR scanner by subjects with both significant motor and cognitive impairment. There have been a number of fMRI studies examining PD subjects with no cognitive impairment or isolated executive dysfunction [118]; these are beyond the scope of the current review.
A newer fMRI method, termed ‘functional connectivity’, has been developed that has the potential to assess brain activity at rest that is not in response to a predefined task. A resting connectivity study in a small number of PD patients with mild executive deficits showed reduced deactivation of the posterior cingulate cortex and the precuneus regions of the default mode network compared with healthy controls [119]. Two recent PET studies in PD, measuring CBF during cognitive tasks before and after the administration of the dopamine agonist apomorphine and before and after levodopa, suggest possible dopaminergic modulation of the default mode network [120,121].
Table 3 summarizes the major MRI findings in PDD.
Conclusion
Functional and anatomic imaging studies confirm the heterogeneous nature of parkinsonian dementia syndromes. Differences in the degree or rate of degeneration within different brain systems may account for differences in phenotypic features, such as temporal differences, in the clinical course of PDD versus DLB. Imaging studies demonstrate evidence of combined subcortical and cortical processes in parkinsonian dementia. Distinct posterior and anterior cortical changes may also reflect specific pathologies. Unlike early stage PD, where there is evidence of uniform and severe dopaminergic denervation, subcortical and cortical cholinergic denervation is more prominent in patients with PDD and DLB. Advances in MRI techniques as well as amyloid PET imaging allow for improved characterization of specific gray and white matter contributions to parkinsonian dementias, such as comorbid amyloidopathy and neural system-wide degenerations in the white matter and functional brain networks. The multisystem changes in parkinsonian dementia are illustrated in a PDD subject who underwent multitracer PET and MRI (Figure 7).

Figure 7: Multimodal imaging in a single Parkinson’s disease with dementia
subject (upper row) and comparative normal control subject (healthy
control) images (lower row). The multitracer PET studies show significant
nigrostriatal dopaminergic losses (VMAT2-PET), cholinergic denervation (AchEPET),
mildly elevated but abnormal amyloid binding (11C-Pittsburgh compound
B PET), leukoaraiosis (FLAIR-MRI) and microstructural white matter tract changes
(DTI-MRI). The imaging findings illustrate the multifactorial and heterogeneous
nature processes of parkinsonian dementia.
AchE: Acetylcholinesterase; DTI: Diffusion tensor imaging; FLAIR: Fluid-attenuated
inversion recovery; HC: Healthy control; PDD: Parkinson’s disease with dementia;
VMAT 2: Vesicular monoamine transporter 2.
Future perspective
Further studies are needed to determine the differential contribution of heterogeneous patterns of monoaminergic and cholinergic denervation and proteinopathies, such as amyloidopathy, to the cognitive and neurobehavioral phenotype of the Parkinson syndrome. It is conceivable that the multiple lesions seen in different neural systems in parkinsonian dementia may not simply evolve in parallel, but instead may be additive or may potentiate one another in terms of functional expression. There is also a need for the development of new PET ligands that are specific for neurofibrillary tau or a-synuclein protein aggregation, which help to improve our understanding of the overlap between AD-type pathology and dementia in parkinsonian disorders.
New MR sequences, such as DTI and resting state functional connectivity fMRI are expected to demonstrate widespread neurodegeneration in both white and gray matter areas in PDD and DLB that extend far beyond the degeneration of monoaminergic and cholinergic neurons.
Acknowledgements
The authors would like to thank M Müller for his assistance with the manuscript figures.
Financial & competing interests disclosure
The authors have received grant funding from the NIH (RO1 NS24896, AG05133, AG08671 and P01 NS015655), Department of Veterans Affairs, RSNA and the Michael J Fox Foundation, which has supported some of the work reviewed in this manuscript. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.

References
Papers of special note have been highlighted as:
• • of considerable interest
- Mayeux R, Stern Y, Rosenstein R et al. An estimate of the prevalence of dementia in idiopathic Parkinson’s disease. Arch. Neurol. 45, 260–262 (1988).
- Marder K, Tang M-X, Côté L, Stern Y, Mayeux R. The frequency and associated risk factors for dementia in patients with Parkinson’s disease. Arch. Neurol. 52, 695–701 (1995).
- Green J, Mcdonald WM, Vitek JL et al. Cognitive impairments in advanced PD without dementia. Neurology 59, 1320–1324 (2002).
- Aarsland D, Bronnick K, Larsen JP, Tysnes OB, Alves G. Cognitive impairment in incident, untreated Parkinson disease: the Norwegian ParkWest study. Neurology 72(13), 1121–1126 (2009).
- Hely MA, Reid WG, Adena MA, Halliday GM, Morris JG. The Sydney multicenter study of Parkinson’s disease: the inevitability of dementia at 20 years. Mov. Disord. 23(6), 837–844 (2008).
- Mckeith IG, Dickson DW, Lowe J et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65(12), 1863–1872 (2005).
- Aarsland D, Bronnick K, Williams-Gray C et al. Mild cognitive impairment in Parkinson disease: a multicenter pooled analysis. Neurology 75(12), 1062–1069 (2010).
- Williams-Gray CH, Evans JR, Goris A et al. The distinct cognitive syndromes of Parkinson’s disease: 5 year follow-up of the CamPaIGN cohort. Brain 132(Pt 11), 2958–2969 (2009).
- Mattila PM, Roytta M, Torikka H, Dickson DW, Rinne JO. Cortical Lewy bodies and Alzheimer-type changes in patients with Parkinson’s disease. Acta Neuropathol. 95, 576–582 (1998).
- Aarsland D, Perry R, Brown A, Larsen JP, Ballard C. Neuropathology of dementia in Parkinson’s disease: a prospective, communitybased study. Ann. Neurol. 58(5), 773–776 (2005).
- Lieberman A, Dziatolowski M, Kupersmith M et al. Dementia in Parkinson’s disease. Ann. Neurol. 6, 355–359 (1979).
- Choi SA, Evidente VG, Caviness JN et al. Are there differences in cerebral white matter lesion burdens between Parkinson’s disease patients with or without dementia? Acta Neuropathol. 119(1), 147–149 (2010).
- Beyer MK, Aarsland D, Greve OJ, Larsen JP. Visual rating of white matter hyperintensities in Parkinson’s disease. Mov. Disord. 21(2), 223–229 (2006).
- Gibb W. Dementia and Parkinson’s disease. Br. J. Psychiatry 154, 596–614 (1989).
- Mahler ME, Cummings JL. Alzheimer disease and the dementia of Parkinson disease: Comparative investigations. Alzheimer Dis. Assoc. Disord. 4, 133–149 (1990).
- Churchyard A, Lees A. The relationship between dementia and direct involvement of the hippocampus and amygdala in Parkinson’s disease. Neurology 49, 1570–1576 (1997).
- Jellinger KA. Morphological substrates of mental dysfunction in Lewy body disease: an update. J. Neural Transm. 59(Suppl.), 185–212 (2000).
- Jellinger KA. Alpha-synuclein pathology in Parkinson’s and Alzheimer’s disease brain: incidence and topographic distribution – a pilot study. Acta Neuropathol. 106(3), 191–201 (2003).
- Hughes TA, Ross HF, Musa S et al. A 10-year study of the incidence of and factors predicting dementia in Parkinson’s disease. Neurology 54(8), 1596–1602 (2000).
- Kalaitzakis ME, Pearce RK. The morbid anatomy of dementia in Parkinson’s disease. Acta Neuropathol. 118(5), 587–598 (2009).
- Hurtig HI, Trojanowski JQ, Galvin J et al. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology 54(10), 1916–1921 (2000).
- Kovari E, Gold G, Herrmann FR et al. Lewy body densities in the entorhinal and anterior cingulate cortex predict cognitive deficits in Parkinson’s disease. Acta Neuropathol. 106(1), 83–88 (2003).
- Mattila PM, Rinne JO, Helenius H, Dickson DW, Roytta M. Alpha-synucleinimmunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson’s disease. Acta Neuropathol. 100(3), 285–290 (2000).
- Braak H, Rub U, Jansen Steur EN, Del Tredici K, de Vos RA. Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology 64(8), 1404–1410 (2005).
- Ballard C, Ziabreva I, Perry R et al. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology 67(11), 1931–1934 (2006).
- Halliday G, Hely M, Reid W, Morris J. The progression of pathology in longitudinally followed patients with Parkinson’s disease. Acta Neuropathol. 115(4), 409–415 (2008).
- Sabbagh MN, Adler CH, Lahti TJ et al. Parkinson disease with dementia: comparing patients with and without Alzheimer pathology. Alzheimer Dis. Assoc. Disord. 23(3), 295–297 (2009).
- Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK. Striatal beta-amyloid deposition in Parkinson disease with dementia. J. Neuropathol. Exp. Neurol. 67(2), 155–161 (2008).
- Compta Y, Parkkinen L, O’sullivan SS et al. Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: which is more important? Brain 134(Pt 5), 1493–1505 (2011).
- Galpern WR, Lang AE. Interface between tauopathies and synucleinopathies: a tale of two proteins. Ann. Neurol. 59(3), 449–458 (2006).
- Jellinger KA, Paulus W. Clinico-pathological correlations in Parkinson’s disease. Clin. Neurol. Neurosurg. 94(Suppl.), S86–S88 (1992).
- Tsuboi Y, Uchikado H, Dickson DW. Neuropathology of Parkinson’s disease dementia and dementia with Lewy bodies with reference to striatal pathology. Parkinsonism Relat. Disord. 13(Suppl. 3), S221–S224 (2007).
- Johansen KK, White LR, Sando SB, Aasly JO. Biomarkers: Parkinson disease with dementia and dementia with Lewy bodies. Parkinsonism Relat. Disord. 16(5), 307–315 (2010).
- Arendt T, Bigl V, Arendt A, Tennstedt A. Loss of neurons in the nucleus basalis of Meynert in Alzheimer’s disease, paralysis agitans and Korsakoff ’s Disease. Acta Neuropathol. 61, 101–108 (1983).
- Whitehouse PJ, Hedreen JC, White CL, Price DL. Basal forebrain neurons in the dementia of Parkinson disease. Ann. Neurol. 13, 243–248 (1983).
- Candy JM, Perry RH, Perry EK et al. Pathological changes in the nucleus of Meynert in Alzheimer’s and Parkinson’s diseases. J. Neurol. Sci. 59, 277–289 (1983).
- Nakano I, Hirano A. Parkinson’s disease: neuron loss in the nucleus basalis without concomitant Alzheimer’s disease. Ann. Neurol. 5, 415–418 (1984).
- Tagliavini F, Pilleri G, Bouras C, Constantinidis J. The basal nucleus of Meynert in idiopathic Parkinson’s disease. Acta Neurol. Scand. 70, 20–28 (1984).
- Rogers JD, Brogan D, Mirra SS. The nucleus basalis of Meynert in neurological disease: a quantitative morphological study. Ann. Neurol. 17, 163–170 (1985).
- Ruberg M, Rieger F, Villageois A, Bonnet AM, Agid Y. Acetylcholinesterase and butyrylcholinesterase in frontal cortex and cerebrospinal fluid of demented and non-demented patients with Parkinson’s disease. Brain Res. 362, 83–91 (1986).
- Whitehouse PJ, Martino AM, Wagster MV et al. Reductions in [3H]nicotinic acetylcholine binding in Alzheimer’s disease and Parkinson’s disease: an autoradiographic study. Neurology 38(5), 720–723 (1988).
- Aubert I, Araujo D, Cecyre D, Robitaille Y, Gauthier S, Quirion R. Comparative alterations of nicotinic and muscarinic binding sites in Alzheimer’s and Parkinson’s diseases. J. Neurochem. 58, 529–541 (1992).
- Kamagata K, Motoi Y, Hori M et al. Posterior hypoperfusion in Parkinson’s disease with and without dementia measured with arterial spin labeling MRI. J. Magn. Reson. Imaging 33(4), 803–807 (2011).
- Minoshima S, Foster NL, Kuhl DE. Posterior cingulate cortex in Alzheimer’s disease. Lancet 344(8926), 895 (1994).
- Vander Borght T, Minoshima S, Giordani B et al. Cerebral metabolic differences in Parkinson’s and Alzheimer’s disease matched for dementia severity. J. Nucl. Med. 38, 797–802 (1997).
- Yong SW, Yoon JK, An YS, Lee PH. A comparison of cerebral glucose metabolism in Parkinson’s disease, Parkinson’s disease dementia and dementia with Lewy bodies. Eur. J. Neurol. 14(12), 1357–1362 (2007).
- Bohnen NI, Koeppe RA, Minoshima S et al. Cerebral glucose metabolic features of Parkinson disease and incident dementia: longitudinal study. J. Nucl. Med. 52(6), 848–855 (2011).
- Jokinen P, Scheinin N, Aalto S et al. [11C]PIB-, [18F]FDG-PET and MRI imaging in patients with Parkinson’s disease with and without dementia. Parkinsonism Relat. Disord. 16(10), 666–670 (2010).
- Kehagia AA, Barker RA, Robbins TW. Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson’s disease. Lancet Neurol. 9(12), 1200–1213 (2010).
- Sawada H, Udaka F, Kameyama M et al. SPECT findings in Parkinson’s disease associated with dementia. J. Neurol. Neurosurg. Psychiatry 55(10), 960–963 (1992).
- Liu RS, Lin KN, Wang SJ et al. Cognition and 99Tcm-HMPAO SPECT in Parkinson’s disease. Nucl. Med. Commun. 13(10), 744–748 (1992).
- Derejko M, Slawek J, Wieczorek D, Brockhuis B, Dubaniewicz M, Lass P. Regional cerebral blood flow in Parkinson’s disease as an indicator of cognitive impairment. Nucl. Med. Commun. 27(12), 945–951 (2006).
- Colloby SJ, Fenwick JD, Williams ED et al. A comparison of 99mTc-HMPAO SPET changes in dementia with Lewy bodies and Alzheimer’s disease using statistical parametric mapping. Eur. J. Nucl. Med. Mol. Imaging 29(5), 615–622 (2002).
- Defebvre LJ, Leduc V, Duhamel A et al. Technetium HMPAO SPECT study in dementia with Lewy bodies, Alzheimer’s disease and idiopathic Parkinson’s disease. J. Nucl. Med. 40(6), 956–962 (1999).
- Takahashi R, Ishii K, Shimada K, Ohkawa S, Nishimura Y. Hypoperfusion of the motor cortex associated with parkinsonism in dementia with Lewy bodies. J. Neurol. Sci. 288(1–2), 88–91 (2010).
- Hilker R, Thomas AV, Klein JC et al. Dementia in Parkinson disease: functional imaging of cholinergic and dopaminergic pathways. Neurology 65(11), 1716–1722 (2005).
- Burke JF, Albin RL, Koeppe RA et al. Assessment of mild dementia with amyloid and dopamine terminal positron emission tomography. Brain 134(Pt 6), 1647–1657 (2011).
- Alexander GE, Delong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu. Rev. Neurosci. 9, 357–381 (1986).
- van Beilen M, Portman AT, Kiers HA et al. Striatal FDOPA uptake and cognition in advanced non-demented Parkinson’s disease: a clinical and FDOPA-PET study. Parkinsonism Relat. Disord. 14(3), 224–228 (2008).
- Bruck A, Aalto S, Nurmi E, Bergman J, Rinne JO. Cortical 6-[18F]fluoro-L-dopa uptake and frontal cognitive functions in early Parkinson’s disease. Neurobiol. Aging 26(6), 891–898 (2005).
- Moore RY, Whone AL, Brooks DJ. Extrastriatal monoamine neuron function in Parkinson’s disease: an 18F-dopa PET study. Neurobiol. Dis. 29(3), 381–390 (2008).
- Rossi C, Volterrani D, Nicoletti V et al. “Parkinson–dementia” diseases: a comparison by double tracer SPECT studies. Parkinsonism Relat. Disord. 15(10), 762–766 (2009).
- Colloby SJ, Williams ED, Burn DJ, Lloyd JJ, Mckeith IG, O’Brien JT. Progression of dopaminergic degeneration in dementia with Lewy bodies and Parkinson’s disease with and without dementia assessed using 123I-FP-CIT SPECT. Eur. J. Nucl. Med. Mol. Imaging 32(10), 1176–1185 (2005).
- Kuhl DE, Koeppe RA, Fessler JA et al. In vivo mapping of cholinergic neurons in the human brain using SPECT and IBVM. J. Nucl. Med. 35(3), 405–410 (1994).
- Kuhl D, Minoshima S, Fessler J et al. In vivo mapping of cholinergic terminals in normal aging, Alzheimer’s disease, and Parkinson’s disease. Ann. Neurol. 40, 399–410 (1996).
- Bohnen NI, Kaufer DI, Ivanco LS et al. Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch. Neurol. 60(12), 1745–1748 (2003).
- Shimada H, Hirano S, Shinotoh H et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology 73(4), 273–278 (2009).
- Shinotoh H, Namba H, Yamaguchi M et al. Positron emission tomographic measurement of acetylcholinesterase activity reveals differential loss of ascending cholinergic systems in Parkinson’s disease and progressive supranuclear palsy. Ann. Neurol. 46, 62–69 (1999).
- Bohnen NI, Muller ML, Koeppe RA et al. History of falls in Parkinson disease is associated with reduced cholinergic activity. Neurology 73(20), 1670–1676 (2009).
- Marshall GA, Shchelchkov E, Kaufer DI, Ivanco LS, Bohnen NI. White matter hyperintensities and cortical acetylcholinesterase activity in parkinsonian dementia. Acta Neurol. Scand. 113(2), 87–91 (2006).
- Dubois B, Danze F, Pillon B, Cusimano G, Lhermitte F, Agid Y. Cholinergic-dependent cognitive deficits in Parkinson’s disease. Ann. Neurol. 22, 26–30 (1987).
- Dubois B, Pillon B, Lhermitte F, Agid Y. Cholinergic deficiency and frontal dysfunction in Parkinson’s disease. Ann. Neurol. 28, 117–121 (1990).
- Cooper JA, Sagar HJ, Doherty SM, Jordan N, Tidswell P, Sullivan EV. Different effects of dopaminergic and anticholinergic therapies on cognitive and motor function in Parkinson’s disease. A follow-up study of untreated patients. Brain 115, 1701–1725 (1992).
- Bedard MA, Pillon B, Dubois B, Duchesne N, Masson H, Agid Y. Acute and long-term administration of anticholinergics in Parkinson’s disease: specific effects on the subcortico-frontal syndrome. Brain Cogn. 40, 289–313 (1999).
- Bedard MA, Lemay S, Gagnon JF, Masson H, Paquet F. Induction of a transient dysexecutive syndrome in Parkinson’s disease using a subclinical dose of scopolamine. Behav. Neurol. 11, 187–195 (1998).
- Klein JC, Eggers C, Kalbe E et al. Neurotransmitter changes in dementia with Lewy bodies and Parkinson disease dementia in vivo. Neurology 74(11), 885–892 (2010).
- Choi SH, Jung TM, Lee JE, Lee SK, Sohn YH, Lee PH. Volumetric analysis of the substantia innominata in patients with Parkinson’s disease according to cognitive status. Neurobiol Aging doi:10.1016/j. neurobiolaging.2010.11.015 (2011) (Epub ahead of print).
- Klunk WE, Engler E, Nordberg A et al. Imaging brain amyloid in Alzheimer’s disease using the novel positron emission tomography tracer, Pittsburgh Compound-B. Ann. Neurol. 55, 306–319 (2004).
- Kung MP, Zhuang ZP, Hou C, Kung HF. Development and evaluation of iodinated tracers targeting amyloid plaques for SPECT imaging. J. Mol. Neurosci. 24(1), 49–53 (2004).
- Rowe CC, Ng S, Ackermann U et al. Imaging beta-amyloid burden in aging and dementia. Neurology 68(20), 1718–1725 (2007).
- Foster ER, Campbell MC, Burack MA et al. Amyloid imaging of Lewy body-associated disorders. Mov. Disord. 25(15), 2516–2523 (2010).
- Villemagne VL, Ong K, Mulligan RS et al. Amyloid imaging with 18F-florbetaben in Alzheimer disease and other dementias. J. Nucl. Med. 52(8), 1210–1217 (2011).
- Maetzler W, Reimold M, Liepelt I et al. [11C]PiB binding in Parkinson’s disease dementia. Neuroimage 39(3), 1027–1033 (2008).
- Edison P, Rowe CC, Rinne JO et al. Amyloid load in Parkinson’s disease dementia and Lewy body dementia measured with [11C]PiB positron emission tomography. J. Neurol. Neurosurg. Psychiatry 79(12), 1331–1338 (2008).
- Gomperts SN, Rentz DM, Moran E et al. Imaging amyloid deposition in Lewy body diseases. Neurology 71(12), 903–910 (2008).
- Maetzler W, Liepelt I, Reimold M et al. Cortical PiB binding in Lewy body disease is associated with Alzheimer-like characteristics. Neurobiol. Dis. 34(1), 107–112 (2009).
- Burack MA, Hartlein J, Flores HP, Taylor-Reinwald L, Perlmutter JS, Cairns NJ. In vivo amyloid imaging in autopsy-confirmed Parkinson disease with dementia. Neurology 74(1), 77–84 (2010).
- Laakso MP, Partanen K, Riekkinen P et al. Hippocampal volumes in Alzheimer’s disease, Parkinson’s disease with and without dementia, and in vascular dementia: an MRI study. Neurology 46(3), 678–681 (1996).
- Camicioli R, Moore MM, Kinney A, Corbridge E, Glassberg K, Kaye JA. Parkinson’s disease is associated with hippocampal atrophy. Mov. Disord. 18(7), 784–790 (2003).
- Junque C, Ramirez-Ruiz B, Tolosa E et al. Amygdalar and hippocampal MRI volumetric reductions in Parkinson’s disease with dementia. Mov. Disord. 20(5), 540–544 (2005).
- Bouchard TP, Malykhin N, Martin WR et al. Age and dementia-associated atrophy predominates in the hippocampal head and amygdala in Parkinson’s disease. Neurobiol. Aging 29(7), 1027–1039 (2008).
- Bruck A, Kurki T, Kaasinen V, Vahlberg T, Rinne JO. Hippocampal and prefrontal atrophy in patients with early non-demented Parkinson’s disease is related to cognitive impairment. J. Neurol. Neurosurg. Psychiatry 75(10), 1467–1469 (2004).
- Jokinen P, Bruck A, Aalto S, Forsback S, Parkkola R, Rinne JO. Impaired cognitive performance in Parkinson’s disease is related to caudate dopaminergic hypofunction and hippocampal atrophy. Parkinsonism Relat. Disord. 15(2), 88–93 (2009).
- Ramirez-Ruiz B, Marti MJ, Tolosa E et al. Longitudinal evaluation of cerebral morphological changes in Parkinson’s disease with and without dementia. J. Neurol. 252(11), 1345–1352 (2005).
- Burton EJ, Mckeith IG, Burn DJ, Williams ED, O’Brien JT. Cerebral atrophy in Parkinson’s disease with and without dementia: a comparison with Alzheimer’s disease, dementia with Lewy bodies and controls. Brain 127(Pt 4), 791–800 (2004).
- Ramirez-Ruiz B, Marti MJ, Tolosa E et al. Cerebral atrophy in Parkinson’s disease patients with visual hallucinations. Eur. J. Neurol. 14(7), 750–756 (2007).
- Beyer MK, Janvin CC, Larsen JP, Aarsland D. A magnetic resonance imaging study of patients with Parkinson’s disease with mild cognitive impairment and dementia using voxel-based morphometry. J. Neurol.
- Neurosurg. Psychiatry 78(3), 254–259 (2007). 98 Camicioli R, Sabino J, Gee M et al. Ventricular dilatation and brain atrophy in patients with Parkinson’s disease with incipient dementia. Mov. Disord. 26(8), 1443–1450 (2011).
- Hu MT, White SJ, Chaudhuri KR, Morris RG, Bydder GM, Brooks DJ. Correlating rates of cerebral atrophy in Parkinson’s disease with measures of cognitive decline. J. Neural Transm. 108(5), 571–580 (2001).
- Burton EJ, Mckeith IG, Burn DJ, O’Brien JT. Brain atrophy rates in Parkinson’s disease with and without dementia using serial magnetic resonance imaging. Mov. Disord. 20(12), 1571–1576 (2005).
- Baloh RW, Yue Q, Socotch TM, Jacobson KM. White matter lesions and disequilibrium in older people: I. Case–control comparison. Arch. Neurol. 52, 970–974 (1995).
- Gunning-Dixon FM, Raz N. The cognitive correlates of white matter abnormalities in normal aging: a quantitative review. Neuropsychology 14, 224–232 (2000).
- Baezner H, Blahak C, Poggesi A et al. Association of gait and balance disorders with age-related white matter changes: the LADIS study. Neurology 70(12), 935–942 (2008).
- Novak V, Haertle M, Zhao P et al. White matter hyperintensities and dynamics of postural control. Magn. Reson. Imaging 27(6), 752–759 (2009).
- Burton EJ, Mckeith IG, Burn DJ, Firbank MJ, O’Brien JT. Progression of white matter hyperintensities in Alzheimer disease, dementia with lewy bodies, and Parkinson disease dementia: a comparison with normal aging. Am. J. Geriatr. Psychiatry 14(10), 842–849 (2006).
- Lee SJ, Kim JS, Yoo JY et al. Influence of white matter hyperintensities on the cognition of patients with Parkinson disease. Alzheimer Dis. Assoc. Disord. 24(3), 227–233 (2010).
- Slawek J, Wieczorek D, Derejko M et al. The influence of vascular risk factors and white matter hyperintensities on the degree of cognitive impairment in Parkinson’s disease. Neurol. Neurochir. Pol. 42(6), 505–512 (2008).
- Dalaker TO, Larsen JP, Dwyer MG et al. White matter hyperintensities do not impact cognitive function in patients with newly diagnosed Parkinson’s disease. Neuroimage 47(4), 2083–2089 (2009).
- Bowen BC, Block RE, Sanchez-Ramos J et al. Proton MR spectroscopy of the brain in 14 patients with Parkinson disease. Am. J. Neuroradiol. 16(1), 61–68 (1995).
- Summerfield C, Gomez-Anson B, Tolosa E et al. Dementia in Parkinson disease: a proton magnetic resonance spectroscopy study. Arch. Neurol. 59(9), 1415–1420 (2002).
- Griffith HR, den Hollander JA, Okonkwo OC, O’Brien T, Watts RL, Marson DC. Brain N-acetylaspartate is reduced in Parkinson disease with dementia. Alzheimer Dis. Assoc. Disord. 22(1), 54–60 (2008).
- Griffith HR, Den Hollander JA, Okonkwo OC, O’Brien T, Watts RL, Marson DC. Brain metabolism differs in Alzheimer’s disease and Parkinson’s disease dementia. Alzheimers Dement. 4(6), 421–427 (2008).
- Sugihara S, Kinoshita T, Matsusue E, Fujii S, Ogawa T. Usefulness of diffusion tensor imaging of white matter in Alzheimer disease and vascular dementia. Acta Radiol. 45(6), 658–663 (2004).
- Matsui H, Nishinaka K, Oda M, Niikawa H, Kubori T, Udaka F. Dementia in Parkinson’s disease: diffusion tensor imaging. Acta Neurol. Scand. 116(3), 177–181 (2007).
- Bozzali M, Falini A, Cercignani M et al. Brain tissue damage in dementia with Lewy bodies: n in vivo diffusion tensor MRI study. Brain 128(Pt 7), 1595–1604 (2005).
- Wiltshire K, Concha L, Gee M, Bouchard T, Beaulieu C, Camicioli R. Corpus callosum and cingulum tractography in Parkinson’s disease. Can. J. Neurol. Sci. 37(5), 595–600 (2010).
- Logothetis NK. What we can do and what we cannot do with fMRI. Nature 453(7197), 869–878 (2008).
- Antonelli F, Ray N, Strafella AP. Imaging cognitive and behavioral symptoms in Parkinson’s disease. Expert Rev. Neurother. 10(12), 1827–1838 (2010).
- van Eimeren T, Monchi O, Ballanger B, Strafella AP. Dysfunction of the default mode network in Parkinson disease: a functional magnetic resonance imaging study. Arch. Neurol. 66(7), 877–883 (2009).
- Nagano-Saito A, Liu J, Doyon J, Dagher A. Dopamine modulates default mode network deactivation in elderly individuals during the Tower of London task. Neurosci. Lett. 458(1), 1–5 (2009).
- Argyelan M, Carbon M, Ghilardi MF et al. Dopaminergic suppression of brain deactivation responses during sequence learning. J. Neurosci. 28(42), 10687–10695 (2008).
• • Important epidemiological study of Parkinson’s disease (PD) and dementia.
• • Important neuropathology study of PD with dementia in a longitudinal cohort of PD.
• • Study directly comparing dopaminergic versus cholinergic denervation in PD and PD with dementia.
• • First in vivo cholinergic imaging study in PD patients with dementia.