Research Article - Pharmaceutical Bioprocessing (2016) Volume 4, Issue 4
Endoplasmic Reticulum Stress Causes Renal Epithelial Cell Disjunction
- Corresponding Author:
- Dickhout JG
Department of Medicine, Division of Nephrology, McMaster University and St. Joseph’s Healthcare Hamilton, Hamilton, Ontario, Canada
E-mail: jdickhou@stjosham.on.ca
Abstract
Endoplasmic reticulum (ER) stress results from the accumulation of misfolded proteins in the ER. ER stress is associated with acute kidney injury (AKI) of various causes, including two of the major causes of AKI, nephrotoxic drugs and renal ischemia. AKI is a pathology affecting approximately 15% of all hospital stays. When AKI occurs, it results in both significantly increased mortality and increased length of hospital stay. If the patient survives to leave the hospital, it increases the likelihood of developing chronic kidney disease 10-fold, end stage kidney disease 3-fold, and the incidence of premature death 2-fold. An important pathological feature of AKI is the disruption of epithelial cell junctions in the proximal tubule. This loss of epithelial cell junctions disrupts cell polarity preventing the reabsorption of ultrafiltrate components, including sodium, back into the circulation. We hypothesized that ER stress causes epithelial cell disjunction in human proximal tubular cells by trapping the junctional components in the ER and preventing them from reaching the cell surface. ER stress was induced by a variety of stressors including tunicamycin, thapsigargin, and A23187, all of which differ in their mechanism of action. We found that induction of ER stress, by any means, caused epithelial cell disjunction in the renal tubular epithelium. Disjunction was determined by the translocation of β-catenin, which is both a structural component of epithelial cell adherens junctions and a molecular chaperone, from the cell periphery to the perinuclear region. Co-localization experiments found β-catenin to be trapped within the ER.
Keywords
endoplasmic reticulum stress, epithelial junctions, β-catenin, acute kidney injury
Introduction
The renal tubules are comprised of the renal epithelium. These are important structures in the kidney since they act to reabsorb the majority of material in the ultrafiltrate, thereby concentrating the urine for the disposal of metabolic wastes [1]. This process is mediated by active transport of solutes, and as such, the integrity of the renal epithelial cell junctions is critical in maintaining apical-basal polarity mediating this transport [2].
The proteins of the epithelial junctions are synthesized in the endoplasmic reticulum (ER), a key intracellular organelle dedicated to the synthesis, folding, maturation and transport of nascent proteins [3]. ER stress, a feature of many forms of kidney disease, results from the accumulation of misfolded proteins in the ER, which may be due to an unfavorable local environment for protein folding, or due to mutations in a protein that interferes with its proper folding [4]. The accumulation of these unfolded proteins in the ER activate an evolutionarily conserved cellular response known as the unfolded protein response (UPR) [5]. Protein folding chaperones, such as the glucoseregulated protein 78 (GRP78), help prevent the aggregation of unfolded proteins in the ER by aiding in their folding. These molecules are referred to as molecular chaperones. When ER stress is induced by drugs that disrupt protein folding in the ER, or pathophysiological states that result in protein misfolding, molecular chaperone expression is increased by the UPR to aid in protein folding [6]. Therefore, the level of expression of these molecular chaperones can act as a marker of ER stress. Since GRP78 is a wellestablished molecular chaperone, we utilized it as a marker of ER stress and UPR activation in our experiments [7].
ER stress can be induced by a variety of agents differing in their mechanisms of action. For our purposes, we used thapsigargin (Tm), tunicamycin (Tg), and A23187, as these have previously been shown to induce ER stress within renal tubular epithelial cells [8,9]. Tg is a Ca2+ ATPase inhibitor which raises cytosolic calcium concentration by blocking the ability of the cell to pump calcium into the ER, thus causing these stores to become depleted [10]. Tm induces ER stress by preventing N-linked glycosylation in the ER, and has both antibiotic and antiviral properties [10]. Our group and several others have shown its effects on the kidney, including up regulated ER stress response proteins, extensive tubular interstitial damage at the cortico-medullary junction, and increased apoptosis [7,11-13]. The calcium ionophore A23187 has been shown to induce ER stress by disrupting calcium homeostasis [10].
Acute kidney injury (AKI) is a disease process with no known treatment that may be caused by ER stress [14]. AKI increases patient morbidity and mortality, is commonly found in the hospital setting, and is increasing in incidence [14]. Strategies to diagnose AKI in its early stages and interventions to prevent poor patient outcomes are urgently needed [15]. A major component of AKI is acute tubular necrosis (ATN) [7], which leads to nephron loss through epithelial cell disjunction. Therefore, preservation of epithelial cell junctions may prevent, or reduce, the severity of AKI.
Disturbances in ER homeostasis prevent proteins manufactured in the ER from attaining their proper tertiary structure, and from properly functioning within the cell. Such proteins include important junctional proteins such as E-cadherin and its specific chaperone β-catenin, which interact with the actin cytoskeleton to form the adherens junctions between epithelial cells [2]. β-catenin associating with E-cadherin allows its movement from the ER to the epithelial cell membrane [16]. As such, ER stress induction may interfere with the formation of the E-cadherin and β-catenin junctional complex, leading to epithelial cell disjunction in the renal tubular epithelium. Conditions responsible for AKI, such as nephrotoxic agents [4,7], or ischemia [17], induce such disturbances in ER homeostasis, thus they are potentially responsible for epithelial cell loss in AKI.
Therefore, we hypothesized that the induction of ER stress causes epithelial cell disjunction in proximal tubular epithelial cells. To test this hypothesis, we examined the epithelial cell junctions in HK-2 cells before and after they were treated with known ER stress inducers Tm, Tg, and A23187. We used the change in location of β-catenin and expression of ER stress marker GRP78 to assess disjunction amongst the epithelial cells.
Method
Cell culture
HK-2 cells were used as a model of renal tubular epithelium. These cells are human proximal tubular epithelial cells that have been transformed with human papilloma virus [18]. Cell growth and culture conditions were described previously [8]. Cells were allowed to form epithelial junctions at 100% confluence before experiments were performed.
Reagents
DMSO (Sigma-Aldrich) was used to dissolve non-water soluble reagents, and as a vehicle control. Tm, Tg, and A23187 were dissolved in DMSO. These reagents and 4’6- diamidino- 2-phenylindole (DAPI) were purchased from Sigma- Aldrich (St. Louis, MO). Paraformaldehyde was obtained as a 4% solution in phosphate buffered saline (BioLynx Inc; Brockville, Canada) and used for fixation.
Lactate dehydrogenase (LDH) assay
Cell injury of in vitro human proximal tubule epithelial cells was evaluated by a LDH release assay (Cytotoxicity Detection Kit, Roche). Percentage cell death was calculated as
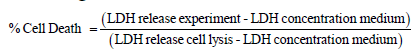
The LDH assay was performed in a medium containing 1% fetal bovine serum. LDH release total cell lysis was determined by lysing cells using 0.1% Triton X-100 in the medium, at the end of the experiment.
Gel electrophoresis
Total cell lysates were generated in 4X SDS lysis buffer with protease inhibitor cocktail added (complete Mini; Roche; Laval, Canada) as previously [8]. To produce equal protein loading, protein levels were determined using a Bio-Rad DC Protein Assay. Electrophoresis separation was performed in a 10% SDS-PAGE reducing gel (Bio-Rad), as described previously [19].
Western blotting
Western blotting was performed using nitrocellulose membranes, and developed with chemiluminescence reagent on high sensitivity X-ray film, as described previously [20].
Fluorescence microscopy
An Olympus IX81 microscope was used for fluorescence microscopy. HK-2 cells were grown on coverslips for immunofluorescent experiments. Cells were stained using anti- GRP78 antibodies (1:100, sc-1050; Santa Cruz Biotechnology), and anti-β-catenin antibody (1:200, no. 2677; Cell Signaling). This was followed by the addition of species-specific secondary antibodies conjugated to an Alexa dye at 488-or 594-nm excitation (1:200; Invitrogen, Molecular Probes). The DNA-specific dye DAPI was used to label the nuclei of cells. PermaFluor (Thermo Scientific) was used to mount the coverslips on microscope slides.
Statistical analysis
Statistics were performed using one-way ANOVA with post-hoc comparisons using Bonferroni’s correction for multiple comparisons of the mean at a 95% confidence level. Values indicate mean ± standard error of the mean.
Results
Cell viability and ER stress induction
The percentage of cell death induced by the various ER stress inducers was determined using a LDH assay kit. The release of the intracellular enzyme LDH into the medium was used as a quantitative measurement of cell viability. LDH assay analysis demonstrated that the doses of the various ER stress inducers resulted in a significant increase in the percentage of cell death (Tm (4.1% ± 0.2%), Tg (3.8% ± 0.2%), and A23187 (3.9% ± 0.3%)), as compared to vehicle (2.3% ± 0.1%). However, when the cell membrane was intentionally disrupted (Lysis) using 0.1% Triton X-100, there was a dramatic, and significant increase in cell death (97.0% ± 0.8%), as determined by the LDH assay (Figure 1). Thus, although ER stress inducers produced a significant increase in cell death, the cell death represented a small percentage of the total number of cells under study.

Figure 1: Percentage of cell death induced by various
ER stressors.
HK-2 cells were treated with vehicle (Veh),
thapsigargin (Tg), tunicamycin (Tm), A23187, and 0.1%
Triton X-100 (Lysis) for 24 h. LDH release was measured
as an indicator of cell toxicity. As intended, Triton X-100
caused 97% cell death. Vehicle had the least amount
of cell death at 2.3%. The other ER stress inducers were
slightly toxic at doses required to induce ER stress but
resulted in relatively little LDH release in comparison to
Lysis. Tg = 200 nM; Tm = 1 μg/mL; A23187 = 5 μM; Triton
X-100 = 0.1% v/v. *, P < 0.05 vs. Veh. ***, P < 0.0001 vs. Veh.
Western blot analysis showed a significant increase in the UPR marker, GRP78, at 24 hours in response to treatment with the ER stressor Tm, indicating ER stress induction (Figure 2A). Cells treated with Tg for 24 hours showed similar results demonstrating a significant increase in expression of the ER stress marker GRP78 (Figure 2B).
• ER stress disrupts renal epithelial cell junctions

Figure 2: Proximal tubular epithelial cells increase expression of GRP78 in response to ER stress inducers.
(A) HK-2 cells were treated with vehicle (Veh) or thapsigargin (Tg; 200 μM) for 18 h. Expression of GRP78 was
significantly increased with Tg treatment. (B) HK-2 cells were treated with vehicle (Veh) or tunicamycin (Tm; 1 μg/ml)
for 24 h. GRP78 levels were significantly increased in response to Tm treatment. *, P < 0.05 vs. Veh.
To demonstrate that ER stress causes epithelial cell disjunction, proximal tubular cells were treated for 24 hours with Tm, Tg, or A23187, and stained for β-catenin (green) and nuclei (blue). Renal proximal tubule epithelial cells treated with DMSO (vehicle) for 24 hours showed β-catenin near the periphery of the cell comprising well-organized epithelial cell junctions (Figure 3). Cells treated with Tg, Tm, or A23187 showed an accumulation of β-catenin in perinuclear region, with reduced levels of β-catenin in the periphery of the cell (Figure 3).
• Co-localization of β-catenin with GRP78 in conditions of ER homeostasis and ER stress
GRP78 employs the KDEL amino acid sequence for ER retention since it is an ER luminal resident protein [21]. As such, GRP78 may be used as a physical marker of the subcellular location of the ER. We utilized confocal microscopy to determine the co-localization of β-catenin with GRP78 during periods of ER homeostasis and during periods of ER stress. Staining of β-catenin (green) is more abundant in the periphery of the cell with vehicle treatment in conditions of ER homeostasis (Figure 4). β-catenin is translocated to the perinuclear region of Tm-, Tg-, and A23187-treated cells, and is co-localized with GRP78 (red), producing yellow regions (Figure 4). Since the ER is located in the perinuclear region, GRP78 and β-catenin staining co-localization in the perinuclear region demonstrates the retention of this epithelial junction protein in the ER of proximal tubule cells undergoing ER stress. This appears to be the mechanism of ER stress mediated epithelial cell disjunction.

Figure 3: ER stress disrupts renal epithelial cell junctions.
Under normal growth conditions, well organized epithelial cell junctions (arrows) are seen in HK-2 cells treated
with vehicle (Veh) for 24 h. Staining shows β-catenin (green) located in the periphery of the renal proximal tubular
epithelial cells. Tg, Tm, and A23187 treatments demonstrate translocation of β-catenin from the periphery of the cell
to the nuclear and perinuclear regions (arrows) after 24 h treatment. Bar = 50 μm. Tg = 200 nM; Tm = 1 μg/ml; A23187
= 5 μM.

Figure 4: GRP78 expression and β-catenin co-localization in response to ER stress.
To determine the effect of ER stress on epithelial cell junctions, HK-2 cells were treated with vehicle (Veh), Tg, Tm,
or A23187 for 24 h. The cells were stained for GRP78 (red), β-catenin (green) and cell nuclei with DAPI (blue). GRP78
upregulation was observed for Tg, Tm, and A23187. GRP78 and β-catenin co-localization indicate failure of junctional
protein β-catenin to travel to cell periphery due to ER retention (yellow, arrows). 40X, Bar = 50 μm. 100X, Bar = 20 μm.
Tg = 200 nM; Tm = 1 μg/mL; A23187 = 5 μM.
Discussion
In these studies, we demonstrated the effect of ER stress induction on epithelial cell adherens junctions. Renal proximal tubule epithelial cells undergo disjunction upon treatment with various ER stress inducers such as Tg, Tm, and A23187, all of which act through different mechanisms. Tg blocks the SERCA pump [23], Tm prevents the N-linked glycosylation of proteins [22], and A23187 is a calcium ionophore [23], all of which disrupt protein trafficking from the ER. Previous studies have been successful in using these agents for the purposes of inducing ER stress within cells [10].
ER stress is known to be a common feature of several renal pathologies [4,24]. The ER is an important organelle that provides a unique environment for normal protein synthesis and folding. Transmembrane, ER luminal resident, and secretory proteins are synthesized in the ER, including proximal tubular cell transporter proteins such as the Na+-K+-ATPase [4]. This protein is critical for the reabsorption of ultrafiltrate components, including sodium [25]. Other important proteins synthesized in the ER include junctional proteins such as E-cadherin and β-catenin [16], which help form adherens junctions between epithelial cells. Maintenance of the integrity of these cell-cell junctions is crucial towards proper kidney function since they help maintain epithelial cell polarity.
The accumulation of misfolded or mutant proteins results in ER stress, and activation of the UPR. The UPR is a coordinated adaptive response designed to improve cellular function and prevent cell death [26,27]. This is achieved through activation of the ER transmembrane sensors PERK, ATF6, and IRE-1 [4]. Under normal conditions, chaperone proteins such as β-catenin facilitate proper protein folding, and formation of junctional complexes between epithelial cells through their association with E-cadherin and the actin cytoskeleton [2]. During conditions of ER stress, β-catenin is released from its E-cadherin anchor in epithelial cell adherens junctions and accumulates in the cytoplasm [8,16]. Our findings demonstrating ER stress-induced epithelial cell disjunction in renal proximal tubule cells is mediated by the retention of β-catenin in the ER. This result is consistent with a study by Ulianich et al [26], where thyroid epithelial cells exhibited loss of organized cell-cell junctions upon treatment with the ER stressors Tm, or Tg. In another study, expression of mutant surfactant protein C resulted in ER stress in type II alveolar epithelial cells. These cells were treated with Tm and showed prominent nuclear localization of β-catenin, as opposed to staining in the cell periphery, as found in vehicle-treated cells [28]. Taken together, available information suggests that disassembly of cell-cell junctions might be part of a common response by epithelial cells to significant or prolonged ER stress. Thus, the condition of ER stress results in epithelial cell disjunction, and loss of epithelial cell polarity.
We have determined, under conditions of ER stress, that β-catenin is retained in the ER. This prevents epithelial junction formation due to the loss of its function as a chaperone and its action to deliver E-cadherin to the epithelial cell membrane, which results in epithelial cell disjunction. These findings have implications in the pathology of AKI since epithelial cell disjunction and loss of cell polarity are related to a rapid and substantial decline in glomerular filtration rate associated with severe AKI. Treatments aimed at preventing epithelial cell disjunction hold promise to alleviate the high morbidity and mortality associated with AKI.
Sources of Funding
Financial support from St. Joseph’s Healthcare Hamilton is acknowledged. Dr. Dickhout holds a Kidney foundation of Canada, Krescent New Investigator award.
Disclosure
Experimental studies were performed by Jasmine Chahal, Ashish Matthews, and Rachel E. Carlisle. Victor Tat and Safaa Naiel assisted with the immunofluorescence staining and with the subsequent imaging process. The article was written by Jasmine Chahal and Jeffrey G. Dickhout. All authors have approved the final copy of this article.
References
- El Kossi M, El Nahas M. Epidemiology and pathophysiology of chronic kidney disease: natural history, risk factors, and management.Comprehensive Clinical Nephrology (3rd edn) Elsevier, Philadelphia. pp: 813-821 (2007).
- Molitoris BA. Therapeutic translation in acute kidney injury: the epithelial / endothelial axis. J Clin Invest. 124, 2355-2363 (2014).
- Deniaud A, Sharaf el dein O, Maillier E, et al. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 27, 285-299 (2008).
- Dickhout JG, Krepinsky JC. Endoplasmic reticulum stress and renal disease. Antioxid Redox Signal. 11, 2341-2352 (2009).
- Wu J, Kaufman RJ. From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 13, 374-384 (2006).
- Dickhout JG, Carlisle RE, Austin RC. Inter-Relationship between Cardiac Hypertrophy, Heart Failure and Chronic Kidney Disease-Endoplasmic Reticulum Stress as a Mediator of Pathogenesis. Circ Res. 108, 629-642 (2011).
- Carlisle RE, Brimble E, Werner KE, et al. 4-Phenylbutyrate inhibits tunicamycin-induced acute kidney injury via CHOP / GADD153 repression. PLoS One. 9, e84663 (2014).
- Carlisle RE, Heffernan A, Brimble E, et al. TDAG51 mediates epithelial-to-mesenchymal transition in human proximal tubular epithelium. Am J Physiol Renal Physiol. 303, F467-481 (2012).
- Nakagawa T, Zhu H, Morishima N, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature.403, 98-103 (2000).
- Dickhout JG, Sood SK, Austin RC. Role of endoplasmic reticulum calcium disequilibria in the mechanism of homocysteine-induced ER stress. Antioxid. Redox Signal. 9, 1863-1873 (2007).
- Marciniak SJ, Yun CY, Oyadomari S, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066-3077 (2004).
- Zinszner H, Kuroda M, Wang X, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982-995 (1998).
- Lhoták S, Sood S, Brimble E, et al. ER stress contributes to renal proximal tubule injury by increasing SREBP-2-mediated lipid accumulation and apoptotic cell death. Am J Physiol Renal Physiol. 303, F266-278 (2012).
- Silver SA, Cardinal H, Colwell K, Burger D, Dickhout JG. Acute kidney injury: preclinical innovations, challenges, and opportunities for translation. Can J Kidney Health Dis. 2, 30 (2015).
- Alge JL, Arthur JM. Biomarkers of AKI: A Review of Mechanistic Relevance and Potential Therapeutic Implications. Clin J Am Soc Nephrol. 10, 147-155 (2015).
- Chen YT, Stewart DB, Nelson WJ. Coupling assembly of the E-cadherin / beta-catenin complex to efficient endoplasmic reticulum exit and basal-lateral membrane targeting of E-cadherin in polarized MDCK cells. J Cell Biol. 144, 687-699 (1999).
- Gao X, Fu L, Xiao M, et al. The nephroprotective effect of tauroursodeoxycholic acid on ischaemia / reperfusion-induced acute kidney injury by inhibiting endoplasmic reticulum stress. Basic Clin Pharmacol Toxicol. 111, 14-23 (2012).
- Ryan MJ, Johnson G, Kirk J, et al. HK-2: an immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Int. 45, 48-57 (1994).
- Dickhout JG, Carlisle RE, Jerome DE, et al. Integrated stress response modulates cellular redox state via induction of cystathionine gamma-lyase: cross-talk between integrated stress response and thiol metabolism. J Biol Chem. 287, 7603-7614 (2012).
- Dickhout JG, Lhoták Š, Hilditch BA, et al. Induction of the unfolded protein response after monocyte to macrophage differentiation augments cell survival in early atherosclerotic lesions. FASEB J. 25, 576-589 (2011).
- Munro S, Pelham HR. A C-terminal signal prevents secretion of luminal ER proteins. Cell.48, 899-907 (1987).
- Yan K, Khoshnoodi J, Ruotsalainen V, Tryggvason K. N-linked glycosylation is critical for the plasma membrane localization of nephrin. J Am Soc Nephrol. 13, 1385-1389 (2002).
- Drummond IA, Lee AS, Resendez E, Steinhardt RA. Depletion of intracellular calcium stores by calcium ionophore A23187 induces the genes for glucose-regulated proteins in hamster fibroblasts. J Biol Chem. 262, 12801-12805 (1987).
- Cybulsky AV. Endoplasmic reticulum stress in proteinuric kidney disease. Kidney Int. 77, 187-193 (2010).
- Rangel LB, Caruso-Neves C, Lara LS, Lopes AG. Angiotensin II stimulates renal proximal tubule Na(+)-ATPase activity through the activation of protein kinase C. Biochim Biophys Acta. 1564, 310-316 (2002).
- Ulianich L, Garbi C, Treglia AS, et al. ER stress is associated with dedifferentiation and an epithelial-to-mesenchymal transition-like phenotype in PC Cl3 thyroid cells. J Cell Sci. 121, 477-486 (2008).
- Dickhout JG, Colgan SM, Lhotak S, Austin RC. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome - A balancing act between plaque stability and rupture. Circulation. 116, 1214-1216 (2007).
- Tanjore H, Cheng DS, Degryse AL, et al. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem. 286. 30972-30980 (2011).